
Abstract
Biological tests performed using 3T3 fibroblasts indicated low cytotoxicities for the complexes mer-[Cr(pic)3] and trans(S,S)-[Cr(Cys)2]−, where pic = picolinate anion and Cys = cysteine. Oxidation of these complexes by hydrogen peroxide was studied in NaOH and NaHCO3 media. Electronic (UV–Vis) and EPR spectroscopies were used to monitor the reaction course. Hydrogen peroxide oxidizes chromium(III) to both [CrV(O2)4]3− and CrVIO4 2− anions in alkaline media and practically completely to CrO4 2− anion in bicarbonate solution. The reactions follow consecutive biphasic or simple first-order kinetics. The first-order decay of [CrV(O2)4]3− anion at pH ≈ 8 was followed by EPR spectroscopy. Based on the obtained kinetic and spectroscopic data, mechanisms for the redox transformations of these chromium(III) complexes are proposed.
Similar content being viewed by others

Introduction
Recently, there has been growing scepticism about the use of chromium(III) pharmaceuticals, mainly due to possible health risks arising from their possible oxidation to chromium(VI) which is believed to induce cancer [1,2,3]. Several reports have demonstrated both extra- and intracellular oxidation of chromium(III) [4,5,6]. The resulting chromate(VI) anion, CrO4 2−, can be transferred into cells via membrane channels suitable for the isostructural anions SO4 2− and HPO4 2− and subsequently converted by cellular reductants into highly reactive chromium(V) and chromium(IV) species which can cause cellular damage, apoptotic body formation and DNA cleavage [7,8,9,10,11,12,13]. On the other hand, it has been suggested that chromium(IV) and chromium(V) play the major role in a recently proposed redox mechanism for the antidiabetic activity of chromium pharmaceuticals [14,15,16,17]. Studies on chromium(III) behaviour in vitro are quite rare; furthermore, its oxidation was carried out either at pH values outside the biological range [18] or using non-biological oxidants such as PbO2 [19]. Partial H2O2 oxidation of chromium(III) in peptide adducts of its dietary supplements in blood serum [20] and partial intracellular chromium(III) oxidation of its nutritional supplement metabolites have been observed [6]. Recently, oxidation of chromium(III) peptides has been examined, and a new fluorescent method of chromium(III) and chromium(VI) determination in cells has been developed [19, 21].
Our tests proved that chromium(III) is not oxidized by H2O2 in NaClO4 and phosphate buffer media within the 7–9 pH range, whereas oxidation does occur in blood plasma at pH 7.4. This phenomenon was the starting point of this project. The work deals with in vitro redox transformation of chromium(III) cysteine and picolinato complexes, trans(S,S)-[Cr(Cys)2]−and mer-[Cr(pic)3], respectively, mediated by hydrogen peroxide which is a biogenic oxidant. The selected complexes are present on the pharmaceutical market as dietary supplements, specifically a source of synthetic biochromium [22,23,24]. The reaction was carried out in media mimicking biological conditions, i.e. in HCO3 − solution of pH ≈ 8 and also in strongly alkaline media. No literature data on kinetics of oxidation of the CrIII–cysteine and CrIII–picolinate complexes by H2O2 have been found. The following problems have focused our attention: (1) the role of HCO3 − medium in the overall reaction, (2) the importance of hydrolytic bond splitting resulting in partial dechelation of the ligands and (3) kinetic and mechanistic differences between reactions of the cysteine and picolinato complexes.
Experimental
Materials
Cysteine, picolinic acid and other chemicals were purchased from Sigma-Aldrich and used without further purification. Na[Cr(Cys)2]·2H2O and [Cr(pic)3]0·H2O were prepared as described in the literature [25, 26]. Chromium was determined spectrophotometrically as CrO4 2− at 372 nm (ɛ 372 = 4830 M−1cm−1) after decomposition of a known amount of the complex in 0.1 M NaOH solution, followed by oxidation of chromium(III) with H2O2. The theoretical chromium contents of Na[Cr(Cys)2] and [Cr(pic)3]0 are 14.9 and 11.9%, respectively; the found values were 14.8 and 11.8%, respectively.
Biological tests
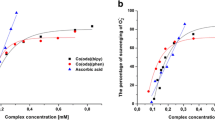
To determine the cytotoxicity of Na[Cr(Cys)2] and [Cr(pic)3]0 aqueous solutions on 3T3 cells, MTT assays were performed. Based on ISO 10993-5 guidelines, Na[Cr(Cys)2] and [Cr(pic)3]0 aqueous solutions showed no cytotoxic activity towards 3T3 cells compared to the control group. Prior to the procedure, 3T3 cells were cultured using DMEM supplemented with 10% FBS and 1% antibiotics and grown at 37 °C, 5% CO2, 99% humidity. The same conditions were applied during the MTT assay, and the investigated dilutions were made using the same culture media. To maintain sterility and ensure purity, both aqueous solutions were filtered using 0.22-μm syringe filters prior to further use. Cells were cultured at 5 × 104 cells per 31.6 mm2 of culture area for 24 h prior to the addition of 10, 5, 1, 0.1% v/v dilutions of the test solutions (4.0 × 10−3 M Na[Cr(Cys)2] and 1.8 × 10−3 M [Cr(pic)3]0), and the MTT assay was performed 24, 48 and 72 h after the addition of the test compound (the MTT assay was performed partially using ISO 10993-5 guidelines (Fig. 1)). Each experimental and control group contained at least eight well replications, and the entire experiment was repeated twice at different time points, yielding the same results. The investigated solutions did not exhibit cytotoxic properties compared to the control group, but cells grown using 5, 1 and 0.1% Na[Cr(Cys)2] exhibited considerably higher proliferation rates at 48 and 72 h after supplementation. Visual analysis using a phase-contrast microscope shows no morphological changes in the studied groups, except for minor morphological changes in cells cultured using 10% Na[Cr(Cys)2] which might result from a high dilution factor of the base medium itself.

Effect of Na[Cr(Cys)2] (Cys) and [Cr(pic)3]0 (pic) on 3T3 cells survivability after 24, 48 and 72 h of treatment. Cell cytotoxicity was determined by MTT assay and expressed as per cent of living cells compared to control group and presented as mean ± SD, *P < 0.05, ANOVA-test and Dunnett’s post hoc versus the control group
Kinetic measurements
The kinetic profiles of the reactions were examined spectrophotometrically using a Hewlett-Packard 8453 diode-array spectrophotometer thermostated with a HP 899090A Peltier temperature controller or using an external Julabo F25 cryostat. All experiments were performed under pseudo-first-order limiting conditions by applying an excess of hydrogen peroxide and sodium hydroxide at 298 K.
The rates of oxidation of both complexes were followed via the absorbance increase over the spectral range 340–420 nm. The reactions were initiated by injection of 0.1 cm3 of the complex solution to 1.9 cm3 of the thermally equilibrated medium solution, prepared in a 1 cm cell directly before the measurement, from appropriate amounts of NaOH or NaHCO3 and H2O2 stock solutions. The concentration of chromium(III) complex was ca. 1.5 × 10−4 M. Kinetics of the reaction was investigated as a function of the hydrogen peroxide concentration (0.1–1.0 M) in 0.6 M NaOH (taking into account its neutralization by H2O2) and bicarbonate concentration (0.3–0.9 M) at 0.2 M H2O2. The sodium hydroxide concentration was accurate to ca. 1% and hydrogen peroxide to ca. 5%. The ionic strength was maintained at 2.0 M (strongly alkaline media) and 1.0 M (bicarbonate solutions) with NaClO4. The pH of the bicarbonate solutions was ca. 8. Additionally, some kinetic traces were recorded within the visible spectral range. The concentration of the soluble Na[Cr(Cys)2] complex was ca. 1 × 10−2 M (1 cm cell). Due to the low solubility of [Cr(pic)3]0, its concentration was only 1 × 10−4 M and the reaction was carried out in a 5 cm cell. Oxidation was initiated by mixing equal volumes of the thermally equilibrated complex and other component solutions.
Each kinetic run was performed for at least three half-lives of the reactions and repeated three times. Absorbance–time changes observed for the cysteine complex reaction obeyed a consecutive first-order reaction model, A → B → C. The data obtained for the [Cr(pic)3]0 complex oxidation were reproduced by a simple A → B reaction model; however, at the highest [H2O2] in NaOH solution a better fit was obtained for the consecutive reaction pattern. The results were independent of the fitting method (multifunctional analyses of the overall spectra using SPECFIT software for a A → B → C reaction model or EnzFitter software for the two separated consecutive reaction steps). The calculated k obs values were independent of the initial complex concentration.
EPR measurements
EPR spectra of reaction mixtures, composed of Na[Cr(Cys)2] or [Cr(pic)2] plus hydrogen peroxide in NaOH or bicarbonate solution, were recorded with a Radiopan EPR SE/X 2541 M spectrometer in X band (ca. 9.33 GHz) with a 100 kHz modulation. The microwave frequency was monitored with a frequency meter. The magnetic field was measured with an automatic NMR-type magnetometer. A flat quartz cell was used. Spectra were recorded at room temperature, and measurements started ca. 180 s after mixing of reagents of the following concentrations: [CrIII] = 1.5–1.8 × 10−4 M, [H2O2] = 0.2 or 0.8 M, [NaOH] = 0.6 M or [NaHCO3] = 0.7 M. The initial intensity of the EPR signal (ca. 180 s after initiation) observed for the reactions carried out in NaOH solution was relatively weak but quickly increased during the reaction course, reaching a maximum at a time dependent mainly on the system studied. For the reaction of the [Cr(Cys)2]− complex in bicarbonate–H2O2 solution, the initial intensity of the EPR signal (ca. 180 s after initiation) was high and then decreased during the reaction course. The collected signal intensity versus time data show a first-order decay of the chromium(V) complex. The product chromium(V) species was identified as [CrV(O2)4]3− based on its EPR parameters, which were identical with those reported in the literature [27, 28]. No EPR signal was registered for the [Cr(pic)3]0 system under the same experimental conditions.
Results and discussion
A large decrease in pH from 13 to 8 results in some important thermodynamic and kinetic consequences for chromium(III)–hydrogen peroxide systems. Both the thermodynamic stability of chromium(III) and redox potential of the H2O2/H2O couple increase with decreasing pH. As a consequence, there is no big change in the thermodynamic driving force of the reaction within the examined pH range. However, our tests showed that the chromium(III) complexes are easily oxidized at pH 13–14 but redox stable at pH 6–8. Addition of NaHCO3 to the system containing hydrogen peroxide and the chromium(III) complex initiates transformation of chromium(III) into CrO4 2− at room temperature. This indicates that several kinetic factors are decisive for the reaction. Firstly, since the pK a value of H2O2 is 11.3, hydrogen peroxide exists in two different protolytic forms in the selected pH ranges: specifically the HO2 − anion and its conjugate acid H2O2 at pH 13–14 and 6–8, respectively. In the presence of HCO3 −, peroxocarbonates are formed. Secondly, in strongly alkaline media the equilibrium between the complex with the open and closed chelate ring is shifted to the latter, which retards an inner sphere electron transfer [25, 26].
Oxidation reactions of the mer-[Cr(pic)3] and trans(S,S)-[Cr(Cys)2]− complexes were monitored spectrophotometrically within the UV–Vis range and by EPR spectroscopy. Experiments were carried out in NaOH and NaHCO3 solutions.
Strongly alkaline media
Isomer mer-[Cr(pic)3] is slowly oxidized by H2O2 at 298 K. Characteristic features of this reaction are shown in Figs. 2 and 3.

Spectral changes observed during the oxidation of [Cr(pic)3]0 by H2O2 in 0.6 M NaOH at 298 K; [CrIII] = 1.5 × 10−4 M, [H2O2] = 0.8 M, l = 1 cm (a) and [CrIII] = 2.0 × 10−3 M, [H2O2] = 0.2 M, l = 5 cm (b), I = 2.0 M, scans every 720 s

Changes of the EPR signal intensity generated during oxidation of [Cr(pic)3]0; [CrIII] = 1.5 × 10−4 M, [H2O2] = 0.8 M, [NaOH] = 0.6 M, I = 2.0 M, T = 298 K
An absorbance increase in the UV region (Fig. 2a) demonstrates accumulation of CrO4 2− accompanied by formation of chromium(V) species, detected as an increase in the characteristic EPR signal (Fig. 3). The presence of chromium(V) is additionally confirmed by a narrow band at 740 nm (Fig. 2b) as observed in other systems [29]. The chromium(V) complex produced spectroscopically is identical to that observed in our previous studies [18, 30, 31] and has been identified as [Cr(O2)4]3−. The red shift of the lower-energy d–d transition band clearly demonstrates hydrolytic transformation of the starting complex to another chromium(III) species as a result of the CrIII–N bond splitting and concomitant chelate ring opening. Comparison of the rate of this process with the rate of chromium(V, VI) formation shows that the latter is preceded by ligand substitution in the starting chromium(III) complex.
Kinetics of the mer-[Cr(pic)3] oxidation have been studied applying large excesses of H2O2 and OH−. In most cases the reaction rate was determined from absorption data in the UV region (CrO4 2− generation). In a few experiments, rate measurements were based on changes in the visible range decay of the starting complex and also as an increase in the EPR signal intensity [chromium(V) formation]. All the methods gave close values of the pseudo-first-order rate constants for the reaction studied:
The overall kinetic traces were reproduced very well by applying a simple A → B reaction model. The consecutive reaction model gave a better fit only at the highest H2O2 concentration used. The results are presented in Table 1.
The data given in Table 1 demonstrate several characteristics for the mer-[Cr(pic)3] oxidation. First, the value of the rate constant for decay of the starting complex, at 0.26 × 10−3 s−1, is not much different than that for the CrO4 2− anion formation (0.41 × 10−3 s−1). Values of the rate constant for the picolinato chelate ring opening determined before, extrapolated to 298 K, vary between 0.7–1.0 × 10−3 s−1 at 0.2−0.9 M Na [26] and are very close to those shown in Table 1. Thus, the rates of CrIII → CrVI transformation and the chelate ring opening are comparable. The rate constant for the second oxidation step is ca. 80 times lower than that for the first one, and the pseudo-first-order rate constant, k obs1, is linearly dependent on the hydrogen peroxide concentration (Fig. 4).

Dependence of k obs1 on [HO2 −] for the [Cr(pic)3]0 oxidation; [OH−] = 0.6 M, T = 298 K, I = 2.0 M
Based on the obtained results, the reaction mechanism shown in Scheme 1 is proposed.

Proposed mechanism for oxidation of mer-[Cr(pic)3]0 by hydrogen peroxide in NaOH solution
The hydrolytic CrIII–pic chelate ring opening plays a key role in the redox process of the mer isomer (Scheme 1). The mechanism of base hydrolysis of mer-[Cr(pic)3]0 was presented in our previous work [26], in which subsequent entry of a water molecule (not OH− ion) into the chromium(III) coordination sphere precedes formation of the hydroxido complexes. In the next steps mono- and dihydroxido derivatives of the starting complex, S–OH and S–(OH)2, are transformed into hydroperoxidochromium(III) intermediates, I–HO2 and I–OH–HO2. The linear dependence of k obs1 on [HO2 −] suggests that the equilibrium involving formation of the intermediate species, I–HO2, is shifted to the left (Fig. 4). Two-electron intramolecular transfer (k et) produces a labile chromium(V) complex which subsequently undergoes fast substitution of the picolinato ligands for peroxido groups. Chromates(VI) are likely formed via chromium(V) disproportionation and further oxidation of chromium(IV) by hydrogen peroxide (not shown in Scheme 1). According to the proposed reaction mechanism, the slope of the plot given in Fig. 4 is interpreted as the product of the rate constant k et and the equilibrium constant K 1 (formation of I–HO2). The hydrolytic chelate ring opening is the rate-determining step of the overall redox process at the highest H2O2 concentration and under these conditions k obs1 = k 1. Deeper insight into the slower reaction stage is impossible due to lack of relevant data.
trans(S,S)-[Cr(Cys)2]−
Some notable differences between oxidation reactions of the cysteine and picolinato complexes were observed, vide Fig. 5.

Spectral changes observed during the oxidation reaction of [Cr(Cys)2]− by 0.2 M H2O2 in 0.6 M NaOH, l = 1 cm; [CrIII] = 1.6 × 10−4 M, scans every 60 s (a); [CrIII] = 1.2 × 10−2 M, scans every 30 s (b); [CrIII] = 5.5 × 10−5 M, spectra 1 and 2 recorded before and immediately after addition of H2O2 (c)
As shown in Fig. 5c, on addition of hydrogen peroxide to the [Cr(Cys)2]− alkaline solution, a practically instantaneous disappearance of the CT absorption band (λ max = 258 nm) characteristic for CrIII–S−II is followed by a substantial absorption increase at λ max = 372 nm confirming accumulation of CrO4 2− (Fig. 5a). Clearly, oxidation of the sulphur atoms precedes oxidation of chromium(III). A shift of the low energy band from 607 to 577 nm (Fig. 5b) shows transformation of the starting complex into another chromium(III) species.
Absorption changes in the visible region are very similar to those observed for base hydrolysis of the cysteine complex [25]. In the first, fast reaction stage, the low energy band shifts towards lower wavelengths and the absorbance slightly increases. The slower subsequent reaction stage is accompanied by a large absorbance decrease and a smaller blue shift. A higher ligand field strength of the generated chromium(III) complexes is consistent with Cr–S bond cleavage and introduction of OH− ligands to the inner coordination sphere. The rate of the subsequent CrIII → CrVI oxidation is not fast enough to prevent accumulation of these chromium(III) species in the system. EPR data recorded during the reaction course demonstrated generation of [CrV(O2)4]3− in parallel with CrO4 2− formation. These data are similar to those presented in Fig. 3 for the mer-[Cr(pic)3]0 isomer. Moreover, the presence of chromium(V) is confirmed by a weak absorption band at λ max 740 nm (Fig. 5b). A simplified reaction course is presented in Scheme 2.

Oxidation of trans(S,S)-[Cr(Cys)2]− by hydrogen peroxide in alkaline medium
In this mechanism, redox transformation of trans(S,S)-[Cr(Cys)2]− by hydrogen peroxide is initiated by rapid oxidation of the sulphur atoms and transformation of the starting complex (CrIII-A) into a CrIII-B species. This reaction is much faster than the subsequent hydrolytic Cr–S bond cleavage. The CrIII-B intermediate (of unknown structure) is hydrolytically transformed into CrIII-C, in which the oxidized ligand is coordinated to chromium(III) by nitrogen and oxygen atoms. Both the CrIII-B and CrIII-C species are oxidized to CrV and CrVI.
Some kinetic data for the trans(S,S)-[Cr(Cys)2]− isomer oxidation are shown in Fig. 5a, b. The reaction obeys two first-order consecutive reaction models, A → B → C. The values of the k obs1 and k obs2 for these two steps, calculated based on (1) visible spectral changes accompanying disappearance of chromium(III) and (2) UV changes accompanying formation of CrO4 2− ion, are convergent to within 10–15% (k obs1) or 5–7% (k obs2). The results are collected in Table 2.
Comparison of the rate constants for hydrolytic Cr–S bond splitting published previously [25] with the rate constants k obs1 and k obs2 gives some insight into the reaction mechanism. The value of the rate constant at 298 K (0.6 M NaOH) for the first Cr–S bond cleavage was found to be 43 × 10−3 s−1 [25], whereas k obs1 increases from 4.4 to 69 × 10−3 s−1 with increasing hydrogen peroxide concentration. Thus, the rate of the oxidation process is slower than the rate of the hydrolytic chelate ring opening, with the exception of the highest oxidant concentration. The rate constants for the second Cr–S bond cleavage and the second oxidation stage are very similar at 298 K (0.6 M NaOH): 1.4 × 10−3 s−1 (hydrolytic Cr–S bond splitting) and 1.1−2.8 × 10−3 s−1 (chromium(III) oxidation). In spite of similarities between the values of the rate constants in these two systems, it should be kept in mind that in the absence of H2O2, the CrIII–S−II bond cleavage takes place, whereas in the presence of H2O2 the CrIII–S−I bond is broken.
The dependence of k obs1 and k obs2 on [HO2 −] is described by two different functions, specifically parabolic (k obs1, Fig. 6) and linear (k obs2, Fig. 7).

Dependence of k obs1 on [HO2 −] for the trans(S,S)-[Cr(Cys)2]− oxidation

Dependence of k obs2 on [HO2 −] for the trans(S,S)-[Cr(Cys)2]− oxidation
The obtained UV–Vis and EPR spectroscopic and kinetic data are consistent with the reaction mechanism presented in a simplified form in Scheme 3.

Proposed mechanism of the oxidation of trans(S,S)-[Cr(Cys)2]−by hydrogen in NaOH
It is assumed that the trans isomer exists in equilibrium with its cis form. The two neighbouring sulphur atoms are then oxidized by hydrogen peroxide, and the S−I–S−I bond is formed to give Sox (where two Cys ligands are transformed into a new ligand Cys’). This oxidation stage results in a rapid disappearance of the CrIII–S−II CT band. The further oxidation steps are preceded by cleavage of the CrIII–S−I bonds, resulting in partial dechelation of the Cys’ ligand and formation of the mono- and dihydroxido derivatives, Sox–OH and Sox–(OH)2. These reaction steps cause changes in the Vis spectrum, namely a blue shift of the lower energy band, characteristic of formation of a new chromium(III) complex with a higher ligand field strength. The Sox–OH and Sox–(OH)2 complexes are reversibly transformed into CrIII–hydroperoxido intermediates: I–HO2 and I–OH–HO2 (K 1, K 2). These equilibria are shifted towards the hydroxo species, which is consistent with the linear k obs2 versus [HO2 −] dependence. The c[HO2 −]2 term, in the expression describing the dependence of k obs1 on [HO2 −], can be rationalized assuming involvement of two HO2 − ions in the synchronic oxidation of CrIII → CrV and S−I → S+I, where one HO2 − ion is in the inner coordination sphere and a second, free HO2 −, interacts with the coordinated sulphur atom from bulk solution. Chromium(III) cannot be transformed into chromium(V) as long as the coordinated sulphur atom is in a negative oxidation state. In the subsequent, faster steps cysteine is oxidized to the hydroxoacid, Cys’’(OH)2, and sulphur to the SO4 2− ion. The final products of chromium(III) oxidation are the metastable [Cr(O2)4]3− complex and CrO4 2− ion, as also proposed for the mer-[Cr(pic)3]0 system. Our suggestions concerning redox transformations of the cysteine ligand are based on the data given in Sharma’s monograph [32] and references therein.
Formation of Sox–OH (k 1) and Sox–(OH)2 (k 2) is the rate-limiting steps for the overall redox process at H2O2 concentrations much higher than those applied. Under the conditions applied in this study, the rate constants of the electron transfer (k et and k et’) and equilibrium constants (K 1 and K 2) determine the rate of chromium(V) and chromium(VI) formation. The slope of the plot in Fig. 7 is the product of the equilibrium constant K 2 and first-order rate constant k et’, whilst the c parameter in the parabolic function (Fig. 6) is the product of the equilibrium constant K 1 and second-order rate constant k et.
Bicarbonate media
Oxidation of chromium(III) complexes in media other than strongly alkaline ones requires the presence of HCO3 − anions. Equilibria in the HCO3 −–H2O2–H2O system are shown in Scheme 4.

Equilibria in the HCO3 −–H2O2–H2O system
No data on the equilibrium constants describing the carbonate–hydrogen peroxide system have been found in the literature. Hence, [H2O2] t and [NaHCO3] t as used in this paper denote the total concentrations of hydrogen peroxide and bicarbonates, independent of their chemical form present in the HCO3 −–H2O2–H2O system (see Scheme 4).
Formation of the final chromium(III) oxidation product, i.e. the CrO4 2− anion, has been monitored from the electronic spectra in the UV region (Figs. 8, 9). The values of the Amax at 372 nm indicate practically complete conversion of the chromium(III) complexes into CrO4 2− ion. No EPR signal was observed during the oxidation process of the mer-[Cr(pic)3]0 isomer over the whole [H2O2] t range applied. In contrast, chromium(V) was detected at the initial stage of the trans(S,S)-[Cr(Cys)2]− oxidation (Fig. 10).

Spectral changes in the UV range during oxidation of mer-[Cr(pic)3]0; [H2O2]t = 0.2 M, [HCO3 −] t = 0.7 M, [CrIII] = 1.2 × 10−4 M, l = 1 cm; scans every 480 s at 298 K

Kinetic traces at 372 nm during oxidation of mer-[Cr(pic)3]0 (a) and trans(S,S)-[Cr(Cys)2]− complexes (b) by H2O2 in bicarbonate media at 298 K; [H2O2]t = 0.2 M, [HCO3 −] t = 0.7 M, [CrIII] = 1.2 × 10−4 M (a) and [CrIII] = 1.7 × 10−4 M (b)

Changes of the EPR signal intensity generated during oxidation of trans(S,S)-[Cr(Cys)2]−; [CrIII] = 1.8 × 10−4 M, [H2O2]t = 0.2 M, [HCO3 −] t = 0.7 M, I = 1.0 M, T = 298 K
The absorbance versus time data collected for the cysteine complex oxidation can be satisfactory reproduced by a double exponential consecutive first-order A → B → C reaction pattern (Fig. 9b), whilst for the picolinato system a good fit was obtained using a simple A → B reaction model (Fig. 9a).
mer-[Cr(pic)3]0
Values of the pseudo-first-order rate constants, k obs, obtained for the mer-[Cr(pic)3]0 isomer oxidation are presented in Table 3.
The data can be rationalized by the simplified reaction mechanism shown in Scheme 5.

Proposed mechanism of the mer-[Cr(pic)3]0 oxidation by H2O2 in bicarbonate media
Chelate ring opening and coordination of a peroxocarbonato anion are essential for the first reaction stage. Formation of an I–HCO4 intermediate is described by the equilibrium constant K 1. The presence of a HCO4 −/CO3 2− ion in the inner coordination sphere of the complex prevents picolinato chelate ring closure. Intramolecular electron transfer from the chromium(III) centre to the coordinated peroxocarbonato ligand produces chromium(V), (k et). Chromium(V) is not accumulated in the system, since protonation of [Cr(O2)4]3− in solutions of pH ~ 8 results in its fast disproportionation or intramolecular redox decomposition. Direct oxidation of chromium(V) by peroxocarbonates cannot be excluded either. The pseudo-first-order rate constant (k obs) is a function of the individual rate constants k 1, k −1, k et plus the equilibrium constant K 1. The limiting case, at very high concentrations of the oxidant and carbonates, considerably simplifies this function to give k obs = k 1. Therefore, under such conditions the chelate ring opening is the rate-controlling step.
Oxidation of trans(S,S)-[Cr(Cys)2]−
An important difference between the picolinato and cysteine complexes is observed in the initial reaction phase. Oxidation of the sulphur atoms, deduced from disappearance of the CT CrIII–S−II bond at 258 nm, is practically instant, as was observed for the reaction in NaOH media (Fig. 5c). An unstable chromium(V) species was then detected during the oxidation process (Fig. 10). It is the same [Cr(O2)4]3− anion as observed in NaOH media. Its concentration reaches a maximum at the very beginning of the reaction and then decreases according to a simple first-order rate law. The obtained value of the pseudo-first-order rate constant for the [Cr(O2)4]3− decay is (6.78 ± 0.26) × 10−3 s−1.
Oxidation of the CrIII-Cys complex by H2O2 in NaHCO3 media follows a biphasic A → B → C course and results in conversion of chromium(III) to CrO4 2−. Values of the pseudo-first-order rate constants for both of the chromium(III) oxidation stages are given in Table 4.
The data in Table 4 show that the second oxidation stage is ca. 10 times slower than the first. Moreover, an increase in bicarbonate concentration accelerates the overall process, as also observed for the mer-[Cr(pic)3]0 system. The values of the k obs1 and k obs2 increase by about 100 and 65%, respectively, within the 0.3–0.9 M (HCO3 −) t range. Rate measurements performed at 0.5 M (HCO3 −) t demonstrated practically the same (within 5%) values of the rate constants within 0.2–0.8 M (H2O2) t .
The proposed reaction sequence is presented in Scheme 6.

Proposed mechanism of the trans(S,S)-[Cr(Cys)2]− oxidation in H2O2–HCO3 − system
In this mechanism, rapid oxidation of the sulphur atoms and formation of an Sox precursor are followed by Cr–S bond cleavage and formation of Sox–H2O (k 1) and Sox–(H2O)2 (k 2) chromium(III) complexes, which subsequently undergo anation by peroxocarbonate to produce I–HCO4 (K 1) and I–H2O–HCO4 (K 2) intermediates. In the subsequent step, electron transfer from chromium(III) to the coordinated peroxocarbonates generates chromium(V). We assume that oxidation of I–HCO4 requires the participation of an additional HCO4 − ion to prevent the reverse CrV → CrIII reduction by coordinated sulphur in a negative oxidation state. The next step of the process is CrV → CrVI transformation, for which two reaction paths are observed. The main path involves rapid decay of chromium(V) not stabilized by peroxido ligands. The minor reaction path proceeds via the rather inert [CrV(O2)4]3− anion (6.8 × 10−3 s−3 at 298 K). This rate constant is ca. twice higher than the rate constant for the second oxidation stage (k obs2). Decay of the [CrV(O2)4]3− anion according to a first-order rate expression implies its intramolecular decomposition. The alternative decay mechanism, i.e. disproportionation, would require a second-order dependence.
Comparison of the rate constants given in Tables 3 and 4 clearly demonstrates a much higher redox stability of the picolinato than the cysteine complex.
Conclusions
Fundamental biological criteria, i.e. low cytotoxicity of the mer-[Cr(pic)3]0 and trans(S,S)-[Cr(Cys)2]−complexes, qualify them as sources of synthetic biochromium. Moreover, the fast hydrolytic Cr–S bond cleavage observed for the cysteine complex generates a chemically and biologically active site in the inner coordination sphere, making possible its interaction with biological targets. However, both of these food supplements are quite rapidly oxidized by hydrogen peroxide, giving CrO4 2− ion in bicarbonate media, thus producing mutagenic and carcinogenic species of chromium in higher oxidation states. Chromium(V) present in bicarbonate media decays with a rate comparable to that of the overall redox process, but its concentration is much lower than that of CrO4 2− during the overall reaction course. The picolinato complex is much more redox resistant than the cysteine one. Oxidation of the chromium(III) centre by hydrogen peroxide proceeds via an inner sphere electron transfer pathway controlled by hydrolytic chelate ring opening.
References
Dillon CT, Lay PA, Bonin AM, Dixon NE, Collins TJ, Kostka KL (1993) Carcinogenesis 14:1875
Bartholomäus R, Irwin JA, Shi L, Smith SM, Levina A, Lay PA (2013) Inorg Chem 52:4282
Lay PA, Levina A (2013) In: Reedijk J, Poeppelmeier K (eds) Comprehensive inorganic chemistry II. Metal carcinogens, vol 3.2, 2nd edn. Elsevier, Amsterdam, pp 835–856
Levina A, Lay PA (2008) Chem Res Toxicol 21:563
Levina A, Mulyani A, Lay PA (2007) In: Vincent JB (ed) The nutritional biochemistry of chromium(III). Redox chemistry and biological activities of chromium(III) complexes. Elsevier, Amsterdam, pp 225–256
Wu LE, Levina A, Harris HH, Cai Z, Lai B, Vogt S, James DE, Lay PA (2016) Angew Chem Int Ed 55:1742
Chiu A, Shi XI, Lee WKP, Hill R, Wakeman TP, Katz A, Xu B, Dalal NS, Robertson JD, Chen C, Chiu N, Donehower L (2010) J Environ Sci Health, Part C 3:188
Vincent JB (2010) Dalton Trans 39:3787
Marin R, Ahuja Y, Bose RN (2010) J Am Chem Soc 132:10617
Myers JM, Antholine WE, Myers CR (2011) Toxicology 281:37
Asatiani N, Abuladze M, Kartvelishvili T, Kulikova N, Asanishvili L, Holman H, Sapojikova N (2010) Biometals 23:161
Wise SS, Holmes AL, Qin Q, Xie H, Katsifis SP, Douglas Thompson W, Wise JP (2010) Chem Res Toxicol 23:365
Vincent JB (2007) The nutritional biochemistry of chromium(III). Elsevier, Amsterdam, p 207
Wang ZQ, Cefalu WT (2010) Curr Diabetes Rep 10:145
Cefalu WT et al (2010) Metab Clin Exp 59:755
Abdul-Ghani MA, DeFronzo RA (2008) Oxidative stress in aging. Springer, Berlin, p 191
Ceriello A, Testa R (2009) Diabetes Care 32:232
Kiersikowska E, Kita E, Kita P, Wrzeszcz G (2016) Transit Met Chem 41:435
Headlam HA, Lay PA (2016) J Inorg Biochem 162:227
Nguyen A, Mulyani, Levina A, Lay PA (2008) Inorg Chem 47:4299
Hu X, Chai J, Liu Y, Liu B, Yang B (2016) Spectrochim Acta Part A 153:505
Trent LK, Theiding-Cancel D (1995) J Sports Med Phys Fit 35:273
Yeh GY, Eisenberg DM, Kaptchuk TJ, Philips RS (2003) Diabetes Care 26(4):1277
Slesinski RS, Clarke JJ, San RHC, Gudi R (2005) Mutat Res 585:86
Kiersikowska E, Kita E, Kita P (2015) Transition Met Chem 40:427
Marai H, Kita E, Wiśniewska J (2012) Transit Met Chem 37:55
Dalal NS, Millar JM, Jagadeesh MS, Seehra MS (1981) J Chem Phys 74:1916
Zhang L, Lay PA (1998) Inorg Chem 37:1729
Katafias A, Impert O, Kita P, Wrzeszcz G (2004) Transit Met Chem 29:855
Kiersikowska E, Marai H, Wrzeszcz G, Kita E (2013) Transit Met Chem 38:603
Kiersikowska E, Marai H, Mątewska M, Wrzeszcz G, Kita E (2014) Transit Met Chem 39:361
Sharma VK (2013) Oxidation of amino acids, peptides, and proteins. Kinetics and mechanism. Wiley, Hoboken
Acknowledgements
E.N. would like to express her gratitude to National Science Centre for financial support of this project, grant No. 2015/17/N/ST4/04070. Authors would like to express their gratitude to prof. Grzegorz Wrzeszcz for EPR measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nieczyporowska, E., Kita, E., Katafias, A. et al. Oxidation in vitro of chromium(III) dietary supplements mer-[Cr(pic)3] and trans(S,S)-[Cr(Cys)2]− by hydrogen peroxide. Transit Met Chem 42, 623–633 (2017). https://doi.org/10.1007/s11243-017-0168-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-017-0168-2