
Abstract
Purpose
Methotrexate (MTX) causes dose-limiting gastrointestinal toxicity due to exposure of intestinal tissues, and is a substrate of the multidrug resistance-associated protein (MRP) 1. Here we examine the involvement of MRP1, which is reported to be highly expressed in the proliferative crypt compartment of the small intestine, in the gastrointestinal toxicity of MTX.
Methods
MTX was intraperitonealy administered to mrp1 gene knockout (mrp1 (−/−)) and wild-type (mrp1 (+/+)) mice. Body weight, food and water intake were monitored, intestinal histological studies and pharmacokinetics of MTX were examined.
Results
mrp1 (−/−) mice more severely decreased body weight, food and water intake than mrp1 (+/+) mice. Almost complete loss of villi throughout the small intestine in mrp1 (−/−) mice was observed, whereas the damage was only partial in mrp1 (+/+) mice. Plasma concentration and biliary excretion profiles of MTX were similar in mrp1 (−/−) and mrp1 (+/+) mice, though accumulation of MTX in immature proliferative cells isolated from mrp1 (−/−) mice was much higher compared to mrp1 (+/+) mice. Immunostaining revealed localization of Mrp1 in plasma membrane of the intestinal crypt compartment in mrp1 (+/+) mice, but not in mrp1 (−/−) mice.
Conclusion
Mrp1 determines the exposure of proliferative cells in the small intestine to MTX, followed by gastrointestinal toxicity.
Similar content being viewed by others
INTRODUCTION
Methotrexate (MTX) has been widely used for the treatment of childhood acute leukemia and rheumatoid arthritis. The clinical application of this drug is restricted by dose-limiting toxicity: myelosuppression and gastrointestinal toxicity. The former side effect can be ameliorated by coadministration with G-CSF (1). However, although it was reported that the intestinal toxicity may be potentiated by prolonged retention of MTX because of its enterohepatic circulation (2,3), the mechanism(s) involved in the toxicity remain to be fully clarified.
In order to understand the disposition of MTX in the body, the membrane transport process must be considered. MTX is one of those therapeutic agents whose pharmacokinetics is predominantly governed by xenobiotic transporters. For example, both multidrug resistance associated protein 2 (Mrp2/Abcc2) and breast cancer resistance protein (BCRP/ABCG2) are suggested to be involved in the biliary excretion of MTX (4,5), and organic anion transporters (OAT1/SLC22A6 and OAT3/SLC22A8) are proposed to be involved in basolateral uptake of MTX in the kidney (6). However, though several transporters have been accepted to influence MTX disposition, little information is available about the relation between intestinal transporter(s) and the gastrointestinal toxicity of MTX.
The small intestine plays a major role in the absorption of various nutrients, and transporters responsible for the uptake of nutrients, such as glucose, vitamins and oligopeptides, are expressed on apical membranes of epithelial cells. Among them, oligopeptide transporter (PEPT1/SLC15A1) has been proposed to be involved not only in gastrointestinal absorption of di- and tripeptides, but also in that of various therapeutic agents, including ß-lactam antibiotics (7,8). Similarly, both reduced folate carrier (RFC/SLC19A1) and SLC46A1, originally identified as a heme carrier protein (HCP1), were reported to be expressed in small intestine, and both recognize folate and MTX (9,10). RFC was suggested to be partly involved in the influx of MTX in a rat IEC-6 intestinal epithelial cell line (11), while HCP1/SLC46A1 was recently demonstrated to mediate intestinal uptake of MTX (12).
In addition to such influx transporters, various types of xenobiotic transporters have been suggested to pump out therapeutic agents from the intestinal cells. For example, P-glycoprotein (P-gp/ABCB1) and Mrp2 limit the oral bioavailability of certain ß-blockers and new quinolone antibiotics by extruding these drugs into the intestinal tract (13,14). Both Mrp2 and Mrp3/Abcc3 are expressed in the small intestine, and accept MTX as a substrate (15,16). Thus, the exposure of intestinal cells to MTX probably depends on membrane permeation of MTX mediated by these transporters.
It should be noted, however, that all the transporters mentioned above are localized in the absorptive epithelial cells of the small intestine. Transporter(s) expressed in proliferative cells, such as small intestinal stem cells, which differentiate into absorptive cells, may also play a role in determining exposure to MTX, and consequently the intestinal toxicity of MTX, since mucotitis may be one of the causes of late-onset diarrhea provoked by MTX. In the present study, we focused on the possible role of Mrp1, which accepts MTX as a substrate (16), in the intestinal toxicity of MTX. Mrp1 has been reported to be mainly localized in proliferative cells in crypts (17), and this led us to the hypothesis that Mrp1 is involved in active efflux of MTX from such proliferative cells as a defensive mechanism to protect the cells against toxicity arising from MTX treatment. To test this hypothesis, we compared the intestinal toxicity induced by MTX treatment in wild-type (mrp1 (+/+)) and mrp1 gene knockout (mrp1 (−/−)) mice in vivo. To clarify how Mrp1 is involved in the toxicity, we further analyzed the involvement of Mrp1 in the disposition of MTX.
MATERIALS AND METHODS
Materials and Animals
MTX and diaminobenzidine were purchased from Wako Pure Chemical Industries (Osaka, Japan). Bromodeoxyuridine, anti-BrdU antibody and aminopterin were from Sigma (St. Louis, MO, USA). Anti-rat IgG and anti-mouse IgG antibodies were purchased from Invitrogen (Carlsbad, CA). Anti-MRP1 antibody (MRPr1) was obtained from ALEXIS (Lausen, Switzerland), anti-mouse IgG peroxidase-linked antibody from Amersham (Budkinghamshire, England), anti-Na+/K+-ATPase antibody from Upstate Biotechnology (Lake Placid, NY, USA). [3H]Methotrexate (1.2 TBq/mmol) was from Moravek Biochemicals Inc. (Brea, CA, USA). [14C]Inulin (89 MBq/g) was from PerkinElmer Life Sciences (Boston, MA, USA).
Five- to six-week-old male FVB (mrp1 (+/+)) and FVB/mrp1 (−/−) mice, originally generated by Wijnholds et al. (18), were purchased from CLEA Japan Inc. (Tokyo, Japan) and Taconic (Germantown, NY, USA), respectively. Animal studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals in Takara-machi Campus of Kanazawa University.
Experimental Schedules and Diarrheal Scores
MTX (0, 25, 50 mg/kg/day) was intraperitoneally administered to mice daily for four successive days. Body weight and food intake were measured daily. Diarrhea that started on day 6 was considered to be delayed onset diarrhea. The severity of this delayed onset diarrhea was scored as follows: − normal stools, + wet and unformed stools, ++ wet stools with perianal staining of the coat (19,20). After the consecutive i.p. administration of MTX (0 or 50 mg/kg), blood was sampled from the cervical vein and plasma was obtained by centrifugation at 2,500×g for 10 min at 4°C in a microcentrifuge (Kubota 1700, Kubota, Tokyo, Japan). Aspartate transaminase (AST), alanine transaminase (ALT), blood urea nitrogen (BUN) and creatinine level in plasma (Cre) were determined at day 4. These parameters were determined by Mitsubishi Chemical Medicine (Tokyo, Japan).
Histological Studies and Immunostaining
Mice were euthanized, and the whole intestines were removed and equally divided into upper, middle and lower segments. Each intestinal segment was fixed in 4% paraformaldehyde for 4 h, embedded in paraffin, sectioned and stained on histological slides with H&E or anti-BrdU antibodies. The small intestine was also removed, quickly immersed in liquid nitrogen, and stored at −80°C until use. The frozen tissue was then sectioned with a cryostat, and the sections were mounted on glass slide and fixed in ice-cold acetone for 10 min. They were then incubated with anti-MRP1 antibody (MRPr1) at 4°C overnight and further incubated with secondary antibody for 1 h at room temperature. Finally, they were mounted in VECTASHIELD mounting medium (Vector Laboratories, Burlingame, CA, USA) to fix the sample. The specimens were examined with an Axiovert S 100 microscope (Carl Zeiss, Jena, Germany), and images were captured with an AxioCam (Carl Zeiss).
Pharmacokinetic Studies
Mice were lightly anesthetized with diethylether, and the gall bladder was cannulated with polyethylene tubing (SP10, Natsume, Tokyo, Japan). MTX (50 mg/kg) was intraperitoneally administered. Blood samples (20 µL) were taken at 15, 30, 60, 120, 150 and 180 min after MTX administration from the tail vein of each mouse and centrifuged to obtain plasma. Bile samples were collected in polyethylene tubes at 5, 10, 20, 30, 45, 60, 90, 120 and 150 min after the drug administration.
Non-compartmental parameters, area under the curve (AUC) and area under the moment curve (AUMC), were calculated by the linear trapezoidal method. Apparent total clearance (CLtot/F), the volume of distribution in the terminal phase (V z/F) mean residence time and biliary clearance (CLbile) were obtained from the following equations:
where F is the intraperitoneal bioavailability, k is the elimination rate constant in the terminal phase, X 0–150 is the MTX amount excreted into the bile from time 0 to 150 min and AUC0–150 is the area under the curve from time 0 to 150 min.
Transport Experiments in Isolated Epithelial Cells
Isolation and fractionation of mouse small intestinal epithelial cells followed the reported method and the composition of the transport buffer was also as reported (21,22). Briefly, the small intestine was surgically removed, cooled in ice-cold buffered saline (21), everted over wooden applicator sticks and washed thoroughly in the saline including 0.1% BSA. Stepwise stripping of epithelial cells from the everted intestines was done by gentle agitation in citrate buffer (96 mM NaCl, 1.5 mM KCl, 27 mM sodium citrate, 8 mM KH2PO4, 7 mM glucose, 0.1% BSA, pH 7.3) varying the length of incubation time (fraction #1 and 2) at 37°C. Fraction #3 and 4 were obtained by additional agitation in phosphate buffer containing 1 mM EDTA, 0.5 mM dithiothreitol, 7 mM glucose and 0.1% BSA again varying the length of incubation time at 37°C. Fractions #1 and #4 mainly contained differentiated and undifferentiated cells, respectively (21,22), and this was confirmed by higher alkaline phosphatase (ALP) activity in fraction #1 than that in fraction #4 (21,22). In the present study, we followed the similar methodology and confirmed that ALP activities in fractions #1, #2, #3 and #4 were 100 ± 5.3, 18.1 ± 1.2, 10.8 ± 1.2 and 7.00 ± 0.5% of maximum for mrp1 (+/+) mice, and 100 ± 3.1, 37.8 ± 3.0, 17.7 ± 1.3 and 5.88 ± 0.4% of maximum for mrp1 (−/−) mice, respectively.
Isolated epithelial cells were incubated in the transport buffer containing [3H]MTX and [14C]inulin. At the designated times, aliquots of the mixture were withdrawn, and the cells were separated from the transport medium by centrifugal filtration through a layer of a mixture of silicon oil (SH550; Toray Dow Corning, Tokyo, Japan) and liquid paraffin (Wako Pure Chemicals) with a density of 1.015 on top of 3 M KOH solution. After solubilization of each cell pellet in KOH, the cell lysate was neutralized with HCl. The associated radioactivity was measured with a liquid scintillation counter, LSC-5100 (Aloka, Tokyo, Japan), with Clearsol I (Nacalai Tesque, Inc., Kyoto, Japan) as the scintillation fluid. Cellular protein content was determined according to the method of Bradford by using a Bio-Rad protein assay kit (Hercules, CA, USA) with bovine serum albumin as the standard.
Determination of MTX by HPLC
The HPLC analysis for MTX was performed according to the previous report (23), using a COSMOSIL 5C18-PAQ (150 × 4.6 mm) column (Nacalai Tesque, Kyoto, Japan). The mobile phase was acetonitrile/0.05 M phosphate buffer (pH 6.9; 5:95; v/v) and the flow rate was 1.0 mL/min. The UV detector (UV-2075 Plus, Jasco, Tokyo, Japan) was operated at a wavelength of 303 nm.
Statistical Analysis
Statistical analysis was performed by using Student’s t test with p < 0.05 as the criterion of significance.
RESULTS
Association of mrp1 Gene with Intestinal Toxicity Evoked by MTX
Intestinal toxicity caused by anticancer drugs is characterized by loss of body weight, anorexia and diarrhea. First, monitoring of the body weight was performed after MTX administration. In the control groups, body weight of both mrp1 (+/+) and mrp1 (−/−) mice gradually increased up to day 7 (Fig. 1A). At 25 mg/kg MTX, no change was observed in the body weight, food intake or water intake between mrp1 (+/+) and mrp1 (−/−) mice up to 7 days. However, there was a marked difference between the two strains in the effect of 50 mg/kg of MTX (Fig. 1A): whereas both mrp1 (+/+) and mrp1 (−/−) mice treated with 50 mg/kg MTX lost about 5% of their body weight by day 5, mrp1 (+/+) mice showed a recovery of body weight by day 7, but the body weight of mrp1 (−/−) mice remained decreased (Fig. 1A). Food and water intake of mrp1 (−/−) mice was decreased in accordance with the change in body weight, and the intake after day 4 in mrp1 (−/−) was lower than in mrp1 (+/+)(Fig. 1B, C).

Effect of MTX administration on body weight (A), food (B) and water (C) intake. MTX (0, 25 or 50 mg/kg) was intraperitoneally administered to mrp1 (+/+) (open symbols) and mrp1 (−/−) (closed symbols) mice for 4 days (once daily as shown as arrows in panel A), and body weight, food and water intake were measured daily. Data are expressed as mean ± S.E.M. of five (mrp1 (+/+)) or six (mrp1 (−/−)) mice (A), or mean values (N = 5–6) of two independent experiments (B, C). *p < 0.05, Significantly different from mrp1 (+/+) mice.
On day 7, lethargy, hunched posture and rough coat were observed in all the mrp1 (−/−) mice treated with 50 mg/kg of MTX, but not in the mrp1 (+/+) mice given the same dose (data not shown). All the mrp1 (−/−) mice treated with 50 mg/kg of MTX exhibited mild or severe diarrhea, whereas no diarrhea was observed in mrp1 (+/+) mice (Table I). Thus, intestinal toxicity, characterized by loss of body weight, poor feeding and diarrhea, was much more severe in mrp1 (−/−) mice than in mrp1 (+/+) mice, indicating the critical role of Mrp1 in gastrointestinal toxicity of MTX. However, it would be possible that the back-crossing to FVB was incomplete and that therefore background effects might not be negligible (18).
Plasma levels of AST, ALT, BUN and Cre in both strains treated with MTX were comparable with those in the control groups (Table I), suggesting that the toxicological effects of MTX treatment in both liver and kidney were minimal.
Disposition of MTX After i.p. Administration
It was reported that approximately 60% of i.v. administered [3H]MTX was excreted into urine, with 35% of the dose being excreted into feces in mice (24). In addition, a substantial amount of MTX was excreted into the bile (4,24), and such biliary excretion may affect the accumulation of MTX in the small intestine. Therefore, both the plasma concentration and biliary excretion of MTX were examined. There was no significant difference in the plasma concentration or biliary excretion profile of MTX between mrp1 (+/+) and mrp1 (−/−) mice (Fig. 2). Thus, the difference in intestinal toxicity of MTX between two strains cannot be accounted for by a difference in disposition. The pharmacokinetic parameters were also similar in the two strains (Table II).

Disposition of MTX in mice. Plasma disappearance (A) and biliary excretion (B) of MTX after i.p. administration (50 mg/kg) was examined in mrp1 (+/+) (open symbols) and mrp1 (−/−) (closed symbols) mice. Data are expressed as mean ± S.E.M. of four (mrp1 (+/+)) or three (mrp1 (−/−)) mice. If error bars are not shown, they lie within the symbol.
Histological Changes
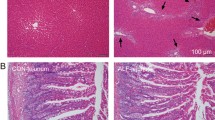
We focused on the localization of Mrp1 in proliferative cells in the small intestine (17), since MTX accumulation in such cells may be responsible for the severe intestinal toxicity observed in mrp1 (−/−) mice. In order to examine this possibility, the architecture of the small intestinal villi and the proliferation of the intestinal cells were investigated. Almost normal structure and BrdU labeling of cells in small intestinal crypts were observed in both mrp1 (+/+) and mrp1 (−/−) mice by day 3 (Fig. 3A). However, the majority of the absorptive cells were severely damaged, with shortened villi and greatly reduced BrdU-labeling of cells in mrp1 (−/−) mice on day 4 (Fig. 3A). Such destruction of small intestinal tissue was not observed in mrp1 (+/+) mice (Fig. 3A). This dramatic difference between mrp1 (+/+) and mrp1 (−/−) mice was also observed in the upper and middle intestines (data not shown). None of MTX-treated mrp1 (−/−) mice died at least for 7 days. HE staining on day 8 revealed proliferation of crypt cells and crypt reconstitution in mrp1 (−/−) (data not shown). MTX withdrawal may thus result in no more damage to intestinal cells, followed by crypt reconstitution. Hence, toxicity observed in mrp1 (−/−) mice may be reversible in the present study.

Histopathological changes in small intestine. MTX (50 mg/kg) was intraperitoneally administered to mrp1 (+/+) and mrp1 (−/−) mice for 4 days. Mice were euthanized on day 2, 3, or 4, and tissue sections from ‘lower’ part of the small intestine (A) and colon (B) were analyzed for morphology with H&E staining (the top panel) and for S-phase cells with immunostaining using anti-BrdU antibody (the bottom panel). Tissue sections from animals without MTX treatment (controls) are also shown. Similar results to those in the ‘lower’ part (A) were also observed in the upper and middle parts of the small intestine (data not shown). Original magnification, ×200.
On the other hand, no prominent damage was observed in the colon of either mrp1 (+/+) or mrp1 (−/−) mice up to 4 days (Fig. 3B), and BrdU labeling of cells in MTX-treated mice was similar to that in control mice (Fig. 3B).
Accumulation of MTX in Intestinal Epithelial Cells
The severe damage in the small intestine and destruction of crypt and villus structure observed in mrp1 (−/−) mice treated with MTX led us to further examine the possibility of excessive MTX accumulation in the proliferative cells of the small intestine in mrp1 (−/−) mice. To examine this possibility, we compared the accumulation of MTX in the isolated intestinal epithelial cells of mrp1 (+/+) and mrp1 (−/−) mice at different stages of maturation. Although biliary excretion of MTX may mainly contribute to MTX accumulation in small intestine, we cannot estimate the actual concentration of MTX in the gastrointestinal tract. Therefore, we first examined the time course of MTX uptake and then measured the steady-state accumulation at various concentrations of MTX outside the epithelial cells (in transport buffer). In fraction #4, which mainly contained undifferentiated cells, steady-state accumulation of MTX (1 µM) were significantly higher in mrp1 (−/−) than in mrp1 (+/+) mice (Fig. 4A). The accumulation of [3H]MTX inside the cells obtained from mrp1 (−/−) mice was much higher at 20 min than that in cells from mrp1 (+/+) mice at all the MTX concentrations examined (Fig. 4A). In contrast, in fraction #1, the accumulation of [3H]MTX in mrp1 (−/−) mice was similar to that in mrp1 (+/+) mice (Fig. 4B).

MTX accumulation in isolated epithelial cells and immunostaining of MRP1 in small intestine. A and B, Intestinal epithelial cells were isolated from mrp1 (+/+) (open bars) and mrp1 (−/−) (closed bars) mice, and fractionated to obtain fractions #4 (A) and #1 (B), which were incubated with the transport buffer (pH 6.0) containing [3H]MTX and [14C]inulin at 37°C for 20 min. C, Immunofluorescence analysis was performed using both anti-MRP1 (green) and anti-Na+/K+ ATPase antibodies (red) in the lower part of small intestine. Colocalization (double staining: yellow) was observed in crypts of mrp1 (+/+) (arrows), but not of mrp1 (−/−) mice. Original magnification, ×200. Inset, Time course of [3H]MTX uptake by fraction #4 obtained from mrp1 (+/+) (open circle) and mrp1 (−/−) (closed circle) mice at 1 µM MTX. Uptake and accumulation were normalized by the medium concentration and represented as cell-to-medium ratio. Data are expressed as mean ± S.E.M. (N = 3–4) *p < 0.05, Significantly different from mrp1 (+/+) mice.
Localization of Mrp1 in Small Intestine
Although specific Mrp1 localization in epithelial cells in crypts has been reported (17), Lorico et al. did not detect Mrp1 in small intestine with western blotting (25). To clarify the localization of Mrp1 in the small intestine, we performed immunohistochemical analysis using anti-Mrp1 antibody in both mrp1 (+/+) and mrp1 (−/−) mice. Na+/K+-ATPase was localized to the basolateral membrane of enterocyte of both mrp1 (+/+) and mrp1 (−/−) mice (Fig. 4C). On the other hand, Mrp1 was detected in plasma membrane in the crypts of mrp1 (+/+) mice, but not mrp1 (−/−) mice (Fig. 4C).
DISCUSSION
Although transporter(s) expressed in differentiated epithelial cells are thought to play a key role in drug absorption (7,8,13–16), little is known about the pharmacological and/or toxicological effects of transporter(s) expressed in undifferentiated cells in the small intestine. The aim of the present study is to clarify whether Mrp1, which was reported to be expressed in proliferative cells of crypts in mice, is involved in the intestinal toxicity provoked by MTX in vivo. We found that mrp1 (−/−) mice given 50 mg/kg MTX showed a decrease of body weight, with decreased intake of food and water (Fig. 1), and developed severe diarrhea (Table I), whereas mrp1 (+/+) mice given the same dose did not show such severe toxic effects (Fig. 1; Table I). These data indicate a critical role of Mrp1 in intestinal toxicity provoked by MTX.
At least three hypotheses could explain the severe gastrointestinal toxicity observed in mrp1 (−/−) mice: (1) Exposure of the small intestine to MTX from either the basolateral or apical side is higher in mrp1 (−/−) mice than in mrp1 (+/+) mice due to the difference in systemic elimination of MTX between the two strains. (2) Accumulation of MTX in the small intestinal cells of mrp1 (−/−) mice is higher than that in mrp1 (+/+) mice, despite the fact that exposure from the extracellular space, i.e., biliary concentration and/or plasma concentration would be the same in the two strains. (3) Mrp1 is involved in some other (unknown) molecular mechanism(s) relevant to MTX toxicity, other than its intrinsic transport function for MTX. The first hypothesis is unlikely because there was no marked difference in the plasma disappearance profile of MTX or in any pharmacokinetic parameter between mrp1 (+/+) and mrp1 (−/−) mice (Table II). The cumulative biliary excretion of MTX was also examined and was slightly higher in mrp1 (−/−) mice than in mrp1 (+/+) mice, but the difference was actually quite small (Fig. 2B). These data suggest that differences in systematic exposure and biliary excretion of MTX cannot explain the difference in the intestinal toxicity of MTX. Biliary excretion of MTX is at least partly mediated by Mrp2, and mRNA for Mrp2 is overexpressed in the liver of mrp1 (−/−) mice (4,5,26). However, such overexpression does not lead to an increase in MTX excretion (Fig. 2B), probably because biliary excretion clearance is close to being plasma-flow-limited (Table II).
The second hypothesis may be supported by the data shown in Figs. 3 and 4. Intestinal stem cells in crypts are thought to proliferate and to differentiate into absorptive cells and goblet cells (27,28). Thus, these proliferative cells are the source of all the differentiated cells, and the death of such proliferative cells may be a critical factor in enteritis induced by MTX. In the present study, on day 4, BrdU-positive cells were greatly reduced in mrp1 (−/−) mice, but this was not the case in mrp1 (+/+) mice, indicating that MTX treatment damaged proliferative cells only in mrp1 (−/−) mice in vivo (Fig. 3A). In addition, Mrp1 is expressed in the membrane of proliferative cells in mrp1 (+/+) mice (Fig. 4C), and in accordance with this, [3H]MTX accumulation in the undifferentiated cells obtained from mrp1 (−/−) mice was much higher than that in the cells from mrp1 (+/+) mice (Fig. 4A). Peng et al. reported that Mrp1 is highly expressed in proliferative cells of crypts in mouse small intestine (17), and this was supported by the immunohistochemical analysis performed in the present study (Fig. 4C). The specificity of the immunohistochemical analysis in the present study was supported in the finding that almost no staining was seen in mrp1 (−/−) mice (Fig. 4C). Therefore, the severe toxicity observed in mrp1 (−/−) mice can be partly explained by deficiency in Mrp1-mediated MTX-extruding activity in mrp1 (−/−) mice, resulting in higher accumulation of MTX inside the proliferative cells. It should also be considered that the fraction of such proliferative cells would be small in the small intestine, and therefore, the MTX concentration observed in the whole intestinal tissues in vivo may not be changed in mrp1 (−/−) mice. We attempted to measure the intestinal MTX concentration after i.p. administration in vivo, but the data exhibited large experimental variation probably because of considerable interindividual variability in biliary excretion and subsequent movement of MTX inside the intestinal lumen (Supplementary figure).
As shown in Fig. 3A, not only the proliferative cells, but also the differentiated absorptive cells localized in small intestinal villi were destroyed by MTX in mrp1 (−/−) mice. This result indicates that our hypothesis, i.e., deficiency in Mrp1-mediated extruding function in proliferative cells, cannot entirely explain the whole mechanism of intestinal toxicity observed in mrp1 (−/−) mice. Mechanisms of MTX-induced enteritis have been studied for a long time, and several factors have been proposed: (1) prolonged exposure to small intestine caused by enterohepatic circulation of MTX, (2) production of reactive oxygen species stimulated by MTX, (3) contribution of mucosal immune cells to the MTX-induced damage and (4) protection of crypt and villus epithelium associated with Peyer’s patches against the MTX-induced damage and (5) mucositis prevention exerted by mucin, the structural component of mucus layer (2,3,29–32). These factors may also contribute to the onset and development of gastrointestinal toxicity of MTX.
Previous studies have demonstrated the increased gastrointestinal toxicity provoked by the administration of etoposide and PMEA in mrp1 (−/−) and mrp4 (−/−) mice, respectively (33,34). Although they described that the damage to stem cells in basolateral epithelium caused oropharyngeal mucosal injury (33), comparison of the drug accumulation between wild-type and knock out mice was not investigated. In the present study, intestinal cells were successfully fractionated, and the higher accumulation of an anticancer drug in proliferative cells of mrp1 (−/−) mice was first demonstrated as one of the possible causes for the higher gastrointestinal toxicity. Important keys to understand the mechanism of diarrhea provoked by anticancer drugs may thus include the concentration of drug in intestinal cells, which would be affected by various transporters including Mrp1.
In addition to Mrp1, Mrp2, Mrp3, Mrp4, P-gp, Bcrp, RFC and HCP are reported to be expressed in the small intestine and accept MTX as a substrate (9,10,15,16,35,36; Fig. 4D). For example, Mrp2 was shown to be localized on apical membranes of epithelial cells, and its expression in upper intestine was higher than that of lower intestine in rats (15). Localization (apical or basal) and regional expression (duodenum, jejunum or ileum) of these transporters would be important factors for MTX disposition in intestines. In spite of so many MTX transporters, however, our present findings indicated extensive accumulation of MTX in undifferentiated cells from mrp1 (−/−) mice (Fig. 4A) and severe gastrointestinal damage observed in mrp1 (−/−) mice (Fig. 3A). Additionally, there is no report about crypt localization of other MTX transporters than Mrp1. These suggest that Mrp1 may control local concentration of MTX in crypt cells, but not absorptive epithelial cells, affecting gastrointestinal toxicity provoked by MTX. However, MTX uptake in fraction #4 exhibited saturation especially for mrp1 (−/−) mice (Fig. 4A), but such saturable uptake was not so clearly observed in fraction #1 (Fig. 4B), implying different influx transporter(s) for MTX between fraction #1 and #4. In addition, if the transporters other than Mrp1 were compensatory up/down-regulated in the crypt cells due to the lack of Mrp1 in mrp1 (−/−) mice, they might also have been involved in the MTX accumulation. Thus, such indirect association between Mrp1 and gastrointestinal toxicity of MTX should also be examined by further analyses.
It would be important to understand whether or not cytotoxic effect of MTX is specific to small intestinal crypt cells. Up to now, except for intestinal crypt cells, there is no information on expression of Mrp1 in undifferentiated cells of other tissues. Etoposide induced damage to the mucosa of the oropharyngeal cavity in mrp1 (−/−) mice (32). Additionally, MTX often causes stomatitis as gastric toxicity. Thus, Mrp1 expressed in undifferentiated cells of oropharyngeal mucosal layer may be also involved in toxicity of MTX.
Though severe damage was found in the small intestine, almost normal structure was observed in the colon of both mice (Fig. 3). This was unexpected, because of Mrp1 is expressed in the colon (17). However, fewer proliferative cells were found in the colon of controls (Fig. 3B), and this may be one of the reasons for insensitivity of the colon to MTX treatment. Another possible reason is that MTX retention in the gastrointestinal tract is thought to be related to its enterohepatic circulation (2,3).
CONCLUSION
We have obtained the evidence that Mrp1 plays a critical role in the gastrointestinal toxicity, especially diarrhea, induced by MTX. In addition to MTX, irinotecan and etoposide often cause severe diarrhea in patients (37,38) and it is noteworthy that all these anticancer drugs are substrates of Mrp1 (25,39). Therefore, Mrp1 would also contribute to diarrhea induced by these anticancer drugs, leading to the necessity of the clinical trial in near future to examine the possible unfavorable effect of MRP1 inhibitors on intestinal toxicity provoked by the administration of the anticancer drugs. Further study is also needed of the effect of genetic and/or functional changes in MRP1 activity on the gastrointestinal toxicity of MTX.
References
K. Welte, M. A. Bonilla, A. P. Gillio, T. C. Boone, G. K. Potter, J. L. Gabrilove, M. A. Moore, R. J. O'Reilly, and L. M. Souza. Recombinant human granulocyte colony-stimulating factor. Effects on hematopoiesis in normal and cyclophosphamide-treated primates. J. Exp. Med. 165:941–948 (1987) doi:10.1084/jem.165.4.941.
S. E. Steinberg, C. L. Campbell, W. A. Bleyer, and R. S. Hillman. Enterohepatic circulation of methotrexate in rats in vivo. Cancer Res. 42:1279–1282 (1982).
D. Griffin, and H. M. Said. The enterohepatic circulation of methotrexate in vivo: inhibition by bile salt. Cancer Chemother. Pharmacol. 19:40–41 (1987) doi:10.1007/BF00296253.
M. Masuda, Y. I’izuka, M. Yamazaki, R. Nishigaki, Y. Kato, K. Ni'inuma, H. Suzuki, and Y. Sugiyama. Methotrexate is excreted into the bile by canalicular multispecific organic anion transporter in rats. Cancer Res. 57:3506–3510 (1997).
P. Breedveld, N. Zelcer, D. Pluim, O. Sönmezer, M. M. Tibben, J. H. Beijnen, A. H Schinkel, O. van Tellingen, P. Borst, and J. H. Schellens. Mechanism of the pharmacokinetic interaction between methotrexate and benzimidazoles: potential role for breast cancer resistance protein in clinical drug-drug interactions. Cancer Res. 64:5804–5811 (2004) doi:10.1158/0008-5472.CAN-03-4062.
M. Takeda, S. Khamdang, S. Narikawa, H. Kimura, M. Hosoyamada, S. H. Cha, T. Sekine, and H. Endou. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J. Pharmacol. Exp. Ther. 302:666–671 (2002) doi:10.1124/jpet.102.034330.
K. Miyamoto, T. Shiraga, K. Morita, H. Yamamoto, H. Haga, Y. Taketani, I. Tamai, Y. Sai, A. Tsuji, and E. Takeda. Sequence, tissue distribution and developmental change in rat intestinal oligopeptide transporter. Biochim. Biophys. Acta. 1305:34–38 (1996).
I. Tamai, T. Nakanishi, K. Hayashi, T. Terao, Y. Sai, T. Shiraga, K. Miyamoto, E. Takeda, H. Higashida, and A. Tsuji. The predominant contribution of oligopeptide transporter PepT1 to intestinal absorption of ß-lactam antibiotics in the rat small intestine. J. Pharm. Pharmacol. 49:796–801 (1997).
Y. Wang, R. Zhao, R. G. Russell, and I. D. Goldman. Localization of murine reduced folate carrier as assessed by immuhistochemical analysis. Biochim. Biophys. Acta. 1513:49–54 (2001) doi:10.1016/S0005-2736(01)00340-6.
M. Shayeghi, G. O. Latunde-Dada, J. S. Oakhill, A. H. Laftah, K. Takeuchi, N. Halliday, Y. Khan, A. Warley, F. E. McCann, R. C. Hider, D. M. Frazer, G. J. Anderson, C. D. Vulpe, R. J. Simpson, and A. T. McKie. Identification of an intestinal heme transporter. Cell. 122:789–801 (2005) doi:10.1016/j.cell.2005.06.025.
Y. Wang, A. Rajgopal, I. D. Goldman, and R. Zhao. Preservation of folate transport activity with a low-pH optimum in rat IEC-6 intestinal epithelial cell lines that lack reduced folate carrier function. Am. J. Physiol. 288:C65–C71 (2005) doi:10.1152/ajpcell.00533.2004.
A. Qiu, M. Jansen, A. Sakaris, S. H. Min, S. Chattopadhyay, E. Tsai, C. Sandoval, R. Zhao, M. H. Akabas, and I. D. Goldman. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 127:917–928 (2006) doi:10.1016/j.cell.2006.09.041.
T. Terao, E. Hisanaga, Y. Sai, I. Tamai, and A. Tsuji. Active secretion of drugs from the small intestinal epithelium in rats by P-glycoprotein functioning as an absorption barrier. J. Pharm. Pharmacol. 48:1083–1089 (1996).
K. Naruhashi, I. Tamai, N. Inoue, H. Muraoka, Y. Sai, N. Suzuki, and A. Tsuji. Involvement of multidrug resistance-associated protein 2 in intestinal secretion of grepafloxacin in rats. Antimicrob. Agents Chemother. 46:344–349 (2002) doi:10.1128/AAC.46.2.344-349.2002.
D. Rost, S. Mahner, Y. Sugiyama, and W. Stremmel. Expression and localization of the multidrug resistance-associated protein 3 in rat small and large intestine. Am. J. Physiol. 28:G720–G726 (2002).
H. Zeng, Z. S. Chen, M. G. Belinsky, P. A. Rea, and G. D. Kruh. Transport of methotrexate (MTX) and folates by multidrug resistance protein (MRP) 3 and MRP1: effect of polyglutamylation on MTX transport. Cancer Res. 61:7225–7232 (2001).
K. C. Peng, F. Cluzeaud, M. Bens, J. P. Van Huyen, M. A. Wioland, R. Lacave, and A. Vandewalle. Tissue and cell distribution of the multidrug resistance associated protein (MRP) in mouse intestine and kidney. J. Histochem. Cytochem. 47:757–767 (1999).
J. Wijnholds, R. Evers, M. R. van Leusden, C. A. Mol, G. J. Zaman, U. Mayer, J. H. Beijnen, M. van der Valk, P. Krimpenfort, and P. Borst. Increased sensitivity to anticancer drugs and decreased inflammatory response in mice lacking the multidrug resistance-associated protein. Nat. Med. 3:1275–1279 (1997) doi:10.1038/nm1197-1275.
A. Kurita, S. Kado, N. Kaneda, M. Onoue, S. Hashimoto, and T. Yokokura. Modified irinotecan hydrochloride (CPT-11) administration schedule improves induction of delayed-onset diarrhea in rats. Cancer Chemother. Pharmacol. 46:211–220 (2000) doi:10.1007/s002800000151.
O. C. Trifan, W. F. Durham, V. S. Salazar, J. Horton, B. D. Levine, B. S. Zweifel, T. W. Davis, and J. L. Masferrer. Cyclooxygenase-2 inhibition with celecoxib enhances antitumor efficacy and reduces diarrhea side effect of CPT-11. Cancer Res. 62:5778–5784 (2002).
F. M. Sirotnak, D. M. Moccio, and C. H. Yang. Similar characteristics of folate analogue transport in vitro in contrast to varying dihydrofolate reductase levels in epithelial cells at different stages of maturation in mouse small intestine. Cancer Res. 44:5204–5211 (1984).
M. M. Weiser. Intestinal epithelial cell surface membrane glycoprotein synthesis. I. An indicator of cellular differentiation. J. Biol. Chem. 248:2536–2541 (1973).
K. Ueda, Y. Kato, K. Komatsu, and Y. Sugiyama. Inhibition of biliary excretion of methotrexate by probenecid in rats: quantitative prediction of interaction from in vitro data. J. Pharmacol. Exp. Ther. 297:1036–1043 (2001).
E. S. Henderson, R. H. Adamson, C. Denham, and V. T. Oliverio. The metabolic fate of tritiated methotrexate 1. Absorption, excretion, and distribution in mice, rats, dogs and monkeys. Cancer Res. 25:1008–1017 (1965).
A. Lorico, G. Rappa, R. A. Finch, D. Yang, R. A. Flavell, and A. C. Sartorelli. Disruption of the murine MRP (multidrug resistance protein) gene leads to increased sensitivity to etoposide (VP-16) and increased levels of glutathione. Cancer Res. 57:5238–5242 (1997).
L. J. Bain, and R. A. Feldman. Altered expression of sulfotransferases, glucuronosyltransferases and mrp transporters in FVB/mrp1-/- mice. Xenobiotica. 33:1173–1183 (2003) doi:10.1080/00498250310001609138.
H. Cheng, and C. P. Leblond. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types. Am. J. Anat. 141:537–561 (1974) doi:10.1002/aja.1001410407.
C. A. Loehry, D. N. Croft, A. K. Singh, and B. Creamer. Cell turnover in the rat small intestinal mucosa: an appraisal of cell loss. Gut. 10:13–16 (1969) doi:10.1136/gut.10.1.13.
I. B. Renes, M. Verburg, N. P. Bulsing, S. Ferdinandusse, H. A. Büller, J. Dekker, and A. W. Einerhand. Protection of the Peyer's patch-associated crypt and villus epithelium against methotrexate-induced damage is based on its distinct regulation of proliferation. J. Pathol. 198:60–68 (2002) doi:10.1002/path.1183.
Y. Miyazono, F. Gao, and T. Horie. Oxidative stress contributes to methotrexate-induced small intestinal toxicity in rats. Scand. J. Gastroenterol. 39:1119–1127 (2004) doi:10.1080/00365520410003605.
B. A. de Koning, J. M. van Dieren, D. J. Lindenbergh-Kortleve, M. van der Sluis, T. Matsumoto, K. Yamaguchi, A. W. Einerhand, J. N. Samsom, R. Pieters, and E. E. Nieuwenhuis. Contributions of mucosal immune cells to methotrexate-induced mucositis. 1. Int. Immunol. 18:941–949 (2006) doi:10.1093/intimm/dxl030.
B. A. de Koning, M. Sluis, D. J. Lindenbergh-Kortleve, A. Velcich, R. Pieters, H. A. Büller, A. W. Einerhand, and I. B. Renes. Methotrexate-induced mucositis in mucin 2-deficient mice. J. Cell. Physiol. 210:144–152 (2007) doi:10.1002/jcp.20822.
J. Wijnholds, G. L. Scheffer, M. van der Valk, P. van der Valk, J. H. Beijnen, R. J. Scheper, and P. Borst. Multidrug resistance protein 1 protects the oropharyngeal mucosal layer and the testicular tubules against drug-induced damage. J. Exp. Med. 188:797–808 (1998) doi:10.1084/jem.188.5.797.
M. G. Belinsky, P. Guo, K. Lee, F. Zhou, E. Kotova, A. Grinberg, H. Westphal, I. Shchaveleva, A. Klein-Szanto, J. M. Gallo, and G. D. Kruh. Multidrug resistance protein 4 protects bone marrow, thymus, spleen, and intestine from nucleotide analogue-induced damage. Cancer Res. 67:262–268 (2007) doi:10.1158/0008-5472.CAN-06-2680.
Y. Mochida, K. Taguchi, S. Taniguchi, M. Tsuneyoshi, H. Kuwano, T. Tsuzuki, M. Kuwano, and M. Wada. The role of P-glycoprotein in intestinal tumorigenesis: disruption of mdr1a suppresses polyp formation in Apc(Min/+) mice. Carcinogenesis. 24:1219–1224 (2003) doi:10.1093/carcin/bgg073.
H. Wang, X. Wu, K. Hudkins, A. Mikheev, H. Zhang, A. Gupta, J. D. Unadkat, and Q. Mao. Expression of the breast cancer resistance protein (Bcrp1/Abcg2) in tissues from pregnant mice: effects of pregnancy and correlations with nuclear receptors. Am. J. Physiol. Endocrinol. Metab. 291:E1295–E1304 (2006) doi:10.1152/ajpendo.00193.2006.
H. Bleiberg. CPT-11 in gastrointestinal cancer. Eur. J. Cancer. 35:371–379 (1999) doi:10.1016/S0959-8049(98)00423-7.
J. T. Hartmann, and H. P. Lipp. Camptothecin and podophyllotoxin derivatives: inhibitors of topoisomerase I and II—mechanisms of action, pharmacokinetics and toxicity profile. Drug Saf. 29:209–230 (2006) doi:10.2165/00002018-200629030-00005.
Z. S. Chen, T. Furukawa, T. Sumizawa, K. Ono, K. Ueda, K. Seto, and S. I. Akiyama. ATP-Dependent efflux of CPT-11 and SN-38 by the multidrug resistance protein (MRP) and its inhibition by PAK-104P. Mol. Pharmacol. 55:921–928 (1999).
Acknowledgement
We thank Ms. Lica Ishida and Mr. Syuichi Yamazaki for technical assistance. This work was supported in part by a Grant-in-Aid for Scientific Research provided by the Ministry of Education, Science and Culture of Japan and funds from the Kanehara Grant Program 2004 of the Ichiro Kanehara Foundation (Tokyo, Japan).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kato, S., Ito, K., Kato, Y. et al. Involvement of Multidrug Resistance-Associated Protein 1 in Intestinal Toxicity of Methotrexate. Pharm Res 26, 1467–1476 (2009). https://doi.org/10.1007/s11095-009-9858-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-009-9858-6