
Abstract
Single crystals of Sn(H2O)3[B10H10] · 3 H2O and Sn(H2O)3[B12H12] · 4 H2O are easily accessible by reactions of aqueous solutions of the acids (H3O)2[B10H10] and (H3O)2[B12H12] with an excess of tin metal powder after isothermal evaporation of the clear brines. Both compounds crystallize with similar structures in the triclinic system with space group P\(\bar{1 }\) and Z = 2. The crystallographic main features are electroneutral \({}_{\infty }^{1} \{\)Sn(H2O)3/1[B10H10]3/3} and \({}_{\infty }^{1} \{\) Sn(H2O)3/1[B12H12]3/3} double chains running along the a-axes. Each Sn2+ cation is coordinated by three water molecules of hydration (d(Sn–O) = 221–225 pm for the B10 and d(Sn–O) = 222–227 pm for the B12 compound) and additionally by hydridic hydrogen atoms of the three nearest boron clusters (d(Sn–H) = 281–322 pm for the B10 and d(Sn–H) = 278–291 pm for the B12 compound), which complete the coordination sphere. Between these tin(II)-bonded water and the three or four interstitial crystal water molecules, classical bridging hydrogen bonds are found, connecting the double chains to each other. Furthermore, there is also non-classical hydrogen bonding between the anionic [BnHn]2− (n = 10 and 12) clusters and the crystal water molecules pursuant to B–Hδ−\(\cdots\)δ+H–O interactions often called dihydrogen bonds.
Similar content being viewed by others
Introduction
Interactions between soft metal cations, e.g. Cu+, Ag+ and Hg2+, and hydro-closo-borate anions [BnHn]2− (n = 10 and 12) were firstly discussed in the 1960s [1]. The salt-like copper(I) compound Cu2[B10H10] shows Cu \(\cdots\) B distances in the range from 214 to 233 pm, indicating a covalent interaction between the Cu+ cation and the hydro-closo-borate anion which was suggested as a three-centered two-electron Cu–H–B bond [2]. This assumption was encouraged by the infrared spectra, which unveils two distinct absorption bands in the B–H stretching area, one for the non-coordinating BH groups and another for the BH groups entailed in the Cu–H–B interactions [3]. Similar can be found in the infrared spectra of the compounds [Cu2(bpa)2B10H10] and [Cu2(bpa)2(OH)2]2[Cu2(B10H10)3] · n CH3CN, both revealing stretching vibrations of BH groups in three-centered Cu–H–B bonds. Additionally the X-ray crystal structure of [Cu2(bpa)2(OH)2B10H10] shows Cu–H contacts in the range of 263–273 pm [4]. For the higher homologous salt-like silver(I) compound Ag2[B10H10] [1, 5], up to now, no crystal structure could be determined, but the presence of three-centered Ag–H–B is supported by infrared spectroscopic data [5]. In the further case of Cu+ and Ag+ cations the review article by Avdeeva et al. [6] should be mentioned, as it provides a good overview of this field.
Up to now, these kind of connections can be found in several other salt-like hydro-closo-borates, especially with cations of the 6th period possessing lone-pair electrons. All of these compounds unveil interesting properties. The thallium(I) salt Tl2[B12H12] [7] exhibits a yellow metal-centered luminescence at room temperature, based on an apparent covalent interaction between Tl+ and the hydridic hydrogen atoms of the [B12H12]2− anions [8]. Besides Tl2[B12H12], there can be also found two lead(II) dodecahydro-closo-dodecaborates, Pb[B12H12] [9] with a pure 6s2 nature of the non-bonding electron lone pair, and Pb(H2O)3[B12H12]·3 H2O [7], where each Pb2+ cation shows an irregular coordination sphere, indicating a lone pair with increased p character and thus stereochemical activity. In the dimeric compound [Pb(bipy)2(B12H12)]2, the lead(II) cations are coordinated by two bipyridyl molecules and via faces to two [B12H12]2− dianions, showing Pb–H contacts in the range of 279–307 pm [10], indicating a 6s2 nature of the non-bonding electron lone pair.
For the homologous decahydro-closo-decaborate anions [B10H10]2−, interactions between these anions and cations with lone-pair electrons were expected. They could be verified through two lead(II) decahydro-closo-decaborates [Pb(H2O)3]2Pb[B10H10]3 · 5.5 H2O and [Pb(H2O)3]Pb[B10H10]2 · 1.5 H2O [11] with different water content, which display hydrated Pb2+ cations each, but also cations only coordinated by hydridic hydrogen atoms of the boron cluster anions, as well as through the complex [Pb(bipy)B10H10] [10, 12]. Herein the Pb2+ cation is surrounded quasi-tetrahedrally by three [B10H10]2− (d(Pb–H) = 266–318 pm) anions and the neutral bipyridyl ligand. As in the lead compound just mentioned above, in the thallium(I) salt Tl2[B10H10] [13] all Tl+ cations are only coordinated by hydridic hydrogen atoms of the boron cluster anions. This compound crystallizes isotypically with the salt-like decahydro-closo-decaborates of some alkali metals (A = Na, K and Rb) [14], but with marked differences in the Tl \(\cdots\) H coordination spheres, based on stereochemically active lone-pair electrons. All these compounds can be seen as potential precursors for metalated boron clusters such as [Et3N][Cu(1-B10H9N2)2] (d(Cu–B) = 218 pm) [15], [P(C6H5)4]2[2-SnCl(C6H5)2B10H9] and [P(C6H5)4]2[2-SnCl2(C6H5)B10H9] (d(Sn–B) = 217–219 pm) [16, 17], eventually leading to features like in the recently published neutral molecule [BiB12H11] with a covalent Bi–B single bond (d(Bi–B) = 230 pm) manifesting an inverted polarity as compared to the typical B–H bonds [18, 19].
Results and Discussion
The newly synthesized triaqua-tin(II) decahydro-closo-decaborate trihydrate crystallizes in the triclinic space group P\(\bar{1 }\) with a = 756.49(5) pm, b = 948.47(6) pm, c = 1034.52(7) pm, α = 69.141(2)°, β = 85.364(2)°, γ = 87.258(2)° as lattice parameters and two formula units per unit cell (Tables 1 and 2) [20].
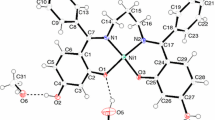
The crystallographically unique Sn2+ cation is coordinated by three oxygen atoms (O1–O3) from the corresponding water molecules of hydration forming the first coordination sphere with d(Sn–O) = 221–225 pm (Fig. 1, left). This coordination polyhedron can be interpreted as non-planar [Sn(H2O)3]2+ cation, structurally very similar to the complex [Pb(H2O)3]2+ cations (d(Pb–O) = 242–252 pm) known from the lead(II) containing hydro-closo-borate hydrates [7, 11]. Furthermore, a second coordination sphere is built up by three decahydro-closo-decaborate anions in such a way, that two of these three [B10H10]2− clusters interact via their triangular faces and a third one via a single corner with each tin(II) cation. Together with the three water molecules of hydration they erect a distorted trigonal antiprism with their barycenters providing for CN = 10 (3 × O + 7 × H) as overall coordination number (Fig. 1, right). This asymmetric coordination of the Sn2+ cation by the three boron clusters allows space for the stereochemically active 5sp lone pair of electrons. The corresponding Sn–H distances range from 281 to 322 pm and are in good accordance with the already known decahydro-closo-decaborates containing lone-pair cations with significant hydrogen interactions, e.g. [Pb(H2O)3]2Pb[B10H10]3 · 5.5 H2O with d(Pb–H) = 249–285 pm [11].

Coordination of the Sn2+ cation by three water molecules of hydration (left) and its second coordination sphere recruited of three additional [B10H10]2− anions (right) in the crystal structure of Sn(H2O)3[B10H10] · 3 H2O
Each [B10H10]2− anion consists of ten independent boron and hydrogen atoms each with B–B distances between 181 and 185 pm within the square antiprism, but 169–171 pm to the apical boron atoms, and B–H distances from 104 to 116 pm. All these distances indicate only minor distortions from the ideal symmetry as bicapped square antiprism, caused by anisotropic direct interactions of three Sn2+ cations attached to each boron cluster. Due to these connections of every [B10H10]2− anion with three tin(II) cations the structure is built up by electroneutral double chains with the formula \({}_{\infty }^{1} \{\)Sn(H2O)3/1[B10H10]3/3} (Fig. 2) running along the a-axis. These double strands are interconnected with each other through three interstitial water molecules (O4w–O6w), which are bonded via classical O–Hδ+\(\cdots\)δ−O hydrogen bonds to the tin(II)-coordinated water molecules of hydration (O1–O3).

Electroneutral double chain with the formula \({}_{\infty }^{1} \{\)Sn(H2O)3/1[B10H10]3/3} in triclinic Sn(H2O)3[B10H10] · 3 H2O propagating parallel to the a-axis as viewed along [010]
The interatomic O \(\cdots\) H distances for these hydrogen bonds range from 171 to 189 pm and can be classified as decently strong, whereas the hydrogen bond interactions among the crystal water molecules O4w and O6w with H42w \(\cdots\) O6w distances of 217 pm are rather weak (Fig. 3, left) [21]. Beneath the classical hydrogen bonds, also unconventional hydrogen-bonds B–Hδ−\(\cdots\)δ+H–O [22] can be observed (Fig. 3, right). These non-classical so-called dihydrogen bonds occur between the negatively polarized hydrogen atoms of the [B10H10]2− anions and the protonic hydrogen atoms of the H2O molecules with d(Hδ−\(\cdots\)δ+H) = 186–225 pm. Each [B10H10]2− anion builds up non-classical hydrogen bonds to the nearest water molecules, represented by O3, O4w, O5w (2 ×) and O6w (2 ×), from which O3 is coordinated directly to a tin(II) cation as water molecule of hydration and the other oxygen atoms stand for free interstitial crystal water molecules (Fig. 4).

Classical (left) and non-classical hydrogen bonds (right) in Sn(H2O)3[B10H10] · 3 H2O

Crystal structure of Sn(H2O)3[B10H10] · 3 H2O as viewed along the a-axis as quasi-hexagonal rod-packing of \({}_{\infty }^{1} \{\)Sn(H2O)3/1[B10H10]3/3} double chains, held together by the interstitial water molecules (H2Ow)
The higher homologous dodecaborate with the composition Sn(H2O)3[B12H12] · 4 H2O crystallizes also in the triclinic space group P\(\bar{1 }\) with a = 741.72(5) pm, b = 991.56(7) pm, c = 1154.63(8) pm, α = 74.471(2)°, β = 83.430(2)°, γ = 87.483(2)° for two formula units per unit cell (Tables 1 and 3) [13]. The unique Sn2+ cation is surrounded by three water molecules of hydration (O1–O3, d(Sn–O) = 222–224 pm) again, leading to the already mentioned [Sn(H2O)3]2+ cation, and three icosahedral [B12H12]2− cluster anions are attached via only one hydrogen atom of each cage (d(Sn–H) = 273–281 pm) providing for a total coordination number of six for the first coordination sphere of the divalent tin (Fig. 5, left). The irregular arrangement of the three oxygen and three hydrogen atoms leaves enough space for a 5sp lone pair of electrons with stereochemical activity. Further tin-hydrogen distances can be found in the range of 315–333 pm, so each of the three boron cages finally coordinates via one triangular face to every Sn2+ cation with a total coordination number of 12 for the first and second coordination sphere (Fig. 5, right).

First (left) and second coordination sphere (right) of the Sn2+ cations in Sn(H2O)3[B12H12] · 4 H2O
A similar structural motif can be found in the orthorhombic lead(II) salt Pb(H2O)3[B12H12] · 3 H2O, also showing stereochemically active lone pairs (6sp) at the Pb2+ cations and direct metal-to-hydrogen interactions (d(Pb–H) = 257–338 pm) [7]. Each quasi-icosahedral [B12H12]2− anion is built up from twelve crystallographically independent boron and hydrogen atoms each (d(B–B) = 177–180 pm, d(B–H) = 106–114 pm) and shows only minor deviations from the ideal values of 178 pm for d(B–B) and 110 pm for d(B–H) for these types of bonds [23]. The pyramidal [Sn(H2O)3]2+ units are interconnected via [B12H12]2− anions to form electroneutral double chains again, now with the formula \({}_{\infty }^{1} \{\)Sn(H2O)3/1[B12H12]3/3}, which run parallel to the a-axis (Fig. 6). These double chains get connected to each other through the four remaining interstitial crystal water molecules (O4w–O7w) erecting the layer structure of Sn(H2O)3[B12H12] · 4 H2O.

View at the electroneutral double chains \({}_{\infty }^{1} \{\)Sn(H2O)3/1[B12H12]3/3} as viewed along the propagating a-axis (left) and along [010] (right) in Sn(H2O)3[B12H12] · 4 H2O
These interstitial crystal water molecules are bonded via strong classical O–Hδ+\(\cdots\)δ−O hydrogen bonds (d(O–H\(\cdots\)O) = 175–221 pm, \(\measuredangle\)(O–H\(\cdots\)O) = 153–174°) to the tin(II)-attached water molecules of hydration (O1–O3; Fig. 7, left). Furthermore, the hydridic hydrogen atoms of the [B12H12]2− anions interact with the protonic hydrogen atoms of the water molecules of hydration. Each [B12H12]2− anion forms six strong non-classical hydrogen bonds with the nearest water molecules, represented by O2, O4w (2 ×), O5w and O6w (2 ×)), with Hδ−\(\cdots\) Hδ+ distances ranging from 191 to 225 pm (Fig. 7, right, and Fig. 8).

Classical hydrogen bonds between two \({}_{\infty }^{1} \{\)Sn(H2O)3/1[B12H12]3/3} chains (left) and the network of non-classical hydrogen bonds (right) in Sn(H2O)3[B12H12] · 4 H2O

View at the triclinic crystal structure of Sn(H2O)3[B12H12] · 4 H2O along [100]
Vibrational Spectroscopy
Despite the two different boron clusters the infrared (IR) and Raman spectra of both compounds are very similar to each other (Fig. 9) and all observed peaks could be assigned successfully. The O–H stretching bands at 3596, 3523 and 3440 cm−1 are broadened due to hydrogen bonds (O–Hδ+\(\cdots\)Oδ−) between the tin(II)-coordinating water molecules of hydration and the free interstitial crystal water molecules.

Vibrational IR and Raman spectra of Sn(H2O)3[B10H10] · 3 H2O (bottom) and Sn(H2O)3[B12H12] · 4 H2O (top)
For the B–H stretching modes between 2400 and 2550 cm−1, there can be found peaks in all four spectra. The sharp peaks in the Raman spectra show no hint for any non-classical hydrogen bonds (B–Hδ−\(\cdots\)δ+H–O), whereas the infrared spectra of Sn(H2O)3[B10H10] · 3 H2O exhibit a band at 2470 cm−1 with a distinct shoulder at 2517 cm−1 and a broad band covering an interval from 2400 to 1800 cm−1. The infrared spectra of Sn(H2O)3[B12H12] · 4 H2O displays a sharp band at 2491 cm−1, with a slight shoulder at 2521 cm−1 and only a weakly broadening between 2390 and 2380 cm−1. The splitting of the B–H stretching mode in a low-frequency and high-frequency region is caused by B–H groups participating in hydrogen bonding (low-frequency shift) and groups that do not participate in hydrogen bonding (high-frequency shift). This clearly signalizing the presence of non-classical hydrogen bonds [24].
Possible occurring B–H\(\cdots\)Sn stretching vibrations are expected in the low field at 2100 to 2000 cm−1 [25, 26], which is debatable for Sn(H2O)3[B10H10] · 3 H2O, but an existing peak might be covered, whereas the IR spectra of Sn(H2O)3[B12H12] · 4 H2O shows no indication of this.
Furthermore, the H–O–H bending vibrations can also be observed in all the spectra. While the IR spectra show sharp bands at 1626 and 1607 cm−1, in the Raman spectra there are just superficial peaks appearing at 1605 and 1602 cm−1. Also the peaks in the O–H bending area reveal a slight broadening in the IR spectra, indicating classical and non-classical hydrogen bonds. The remaining signals encountered in the IR spectra at 1079, 1025, 878 and 713 cm−1 can be assigned to B–B stretching vibrations, also visible in the Raman spectra at 1007, 998, 737, 616 and 580 cm−1. The very intense peaks at 835 and 744 cm−1 are caused by the total symmetric breathing modes of the B10 and B12 cages.
Conclusion
Two tin(II) hydro-closo-borate hydrates were accessible from reactions of elemental tin with aqueous solutions of the acids (H3O)2[B10H10] and (H3O)2[B12H12]. Both compounds crystallize in the same triclinic space group (P\(\bar{1 }\)) with Z = 2 and differ only slightly in structure and composition. In both compounds the Sn2+ cations are coordinated by three oxygen atoms of the corresponding water molecules of hydration, building up non-planar [Sn(H2O)3]2+ units with Sn–O distances in an interval of 221 to 225 pm and a 151 pm off-center shift. Furthermore, three [B10H10]2− and [B12H12]2− cluster anions establish a second coordination sphere with tin(II)-hydrogen contacts (d(Sn–H) = 273–333 pm), providing enough space for the stereochemically active 5sp lone pair of electrons. The Sn2+ cations unveil a coordination behavior very similar to that of the heavier congener Pb2+ in its hydrated hydro-closo-borate compounds. Even the metal hydrogen contacts are in the same range (d(Pb–H) = 249–338 pm) [7, 11]. But in comparison, the Sn–H distances are substantially longer than the Sn–H distance found in the metallacarborane [10-endo-(SnPh3)-10-µ-H-7,8-nido-C2B9H10][trans-Ir(CO)(PPh3)2(MeCN)], with d(Sn–H) = 234 pm [27], as well as the Sn–H contacts found in several dialkylstannylenes (d(Sn–H) = 203–249 pm) [25, 26, 28, 29].
In the same way as the Pb2+ cations, Sn2+ cannot activate the B–H bonds, neither for the [B12H12]2− nor for the more reactive [B10H10]2− case. It seems that the desired B–H bond activation is only provided by Bi3+ cations and [B10H10]2− or [B12H12]2− anions [18, 19] under physiological conditions so far.
Experimental
Sn(H2O)3[B10H10] · 3 H2O
The hydrated decahydro-closo-decaborate compound with divalent tin was yielded after the reaction of tin powder (99.9%, Fluka) with an excess of an aqueous solution of the free acid (H3O)2[B10H10], obtained by passing an aqueous solution of Cs2[B10H10] [30, 31] through a strong acidic ion-exchange column (Merck, Amberlite IR-120). After the hydrogen evolution has concluded, the resulting colorless solution was evaporated at 50 °C to dryness. The white precipitation was again dissolved in water, filtered to remove the remaining tin powder and evaporated isothermally at room temperature to obtain crystals within several days. The pristinely colorless and transparent tin(II) salt crystals with the [B10H10]2− anion, for being a much stronger reductant than [B12H12]2−, becomes brown and opaque over time due to the continuous precipitation of colloidal tin.
Sn(H2O)3[B12H12] · 4 H2O
The analogous [B12H12]2− salt was prepared by the reaction of tin powder (99.9%, Fluka) with an aqueous solution of the free acid (H3O)2[B12H12], obtained by passing an aqueous solution of Cs2[B12H12] [1, 32, 33] through a strong acidic ion-exchange column (Merck, Amberlite IR-120). The reaction solution was heated up to 50 °C for 2 days, filtered and then evaporated at room temperature to yield colorless single crystals within a few days.
An empirical absorption correction was performed by using the program Scalepack [34]. The crystal structure solutions and refinements were carried out with the program package SHELX-97 [35]. Both structure refinements include the positions of all hydrogen atoms without any constraints. The coefficients of the equivalent isotropic displacement parameters are defined as Ueq = 1/3[U11(aa*)2 + U22(bb*)2 + U33(cc*)2 + 2U12aba*b*cosγ + 2U13aca*c*cosβ + 2U23bcb*c*cosα] in pm2 [36]. Further details of the crystal structure investigations may be obtained from the CCDC (www.ccdc.cam.ac.uk) and FIZ Karlsruhe (www.fiz-karlsruhe.de) on quoting the depository numbers 1872762 for Sn(H2O)3[B10H10] · 3 H2O and 1872763 for Sn(H2O)3[B12H12] · 4 H2O.
The infrared spectroscopic data were measured as potassium-bromide powder pellets on a Bruker ALPHA FT/IR spectrometer (Bruker Optik GmbH, Ettlingen, Germany). Raman spectra of the single crystals were recorded by using a XploRA Raman microscope (HORIBA Jobin Yvon GmbH, Bensheim, Germany).
References
E. L. Muetterties, J. H. Balthis, Y. T. Chia, W. H. Knoth, and H. C. Miller (1964). Inorg. Chem. 3, 444.
R. D. Dobrott and W. N. Lipscomb (1962). J. Phys. Chem. 37, 1779.
T. E. Paxton, M. F. Hawthorne, L. D. Brown, and W. N. Lipscomb (1974). Inorg. Chem. 13, 2772.
V. V. Avdeeva, A. E. Dziova, I. N. Polyakova, L. V. Goeva, E. A. Malinina, and N. T. Kuznetsov (2013). Russ. J. Inorg. Chem. 58, 657.
E. A. Malinina, K. Y. Zhizhin, I. N. Polyakova, M. V. Lisovskii, and N. T. Kuznetsov (2002). Russ. J. Inorg. Chem. 47, 1158.
V. V. Avdeeva, E. A. Malinina, I. B. Sivaev, V. I. Bregadze, and N. T. Kuznetsov (2016). Crystals 6, 60.
I. Tiritiris, N.-D. Van, and Th. Schleid (2011). Z. Anorg. Allg. Chem. 637, 682.
H. Kunkely and A. Vogler (2007). Inorg. Chim. Acta 360, 679.
F. M. Kleeberg, L. W. Zimmermann, and Th. Schleid (2014). Z. Anorg. Allg. Chem. 640, 2352.
E. A. Malinina, L. V. Goeva, and N. T. Kuznetsov (2009). Russ. J. Inorg. Chem. 54, 417.
L. W. Zimmermann, F. M. Kleeberg, and Th. Schleid (2017). Z. Anorg. Allg. Chem. 643, 365.
E. A. Malinina, K. A. Solntsev, L. A. Butman, and N. T. Kuznetsov (1989). Koord. Khim. 15, 1039.
F. M. Kleeberg, Doctoral Thesis (University of Stuttgart; Dr. Hut, Munich, 2017).
K. Hofmann and B. Albert (2005). Z. Kristallogr. 220, 142.
L.-L. Ng, B. K. Ng, K. Shelly, C. B. Knobler, and M. F. Hawthorne (1991). Inorg. Chem. 30, 4278.
C. Nachtigal and W. Preetz (1996). Z. Naturforsch. B 51, 1061.
C. Nachtigal and W. Preetz (1997). Z. Naturforsch. B 52, 975.
L. W. Zimmermann, Ng. D. Van, D. Gudat, and Th. Schleid, (2016). Angew. Chem. Int. Ed. 55, 1909.
L. W. Zimmermann, N.-D. Van, D. Gudat, and Th. Schleid (2016). Angew. Chem. 128, 1942.
L. W. Zimmermann, Doctoral Thesis (University of Stuttgart; Dr. Hut, Munich, 2013).
G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond (Oxford University Press, Oxford, 1999).
R. H. Crabtree, P. E. M. Siegbahn, O. Eisenstein, A. L. Rheingold, and T. F. Koetzle (1996). Acc. Chem. Res. 29, 348.
J. A. Wunderlich and W. N. Lipscomb (1960). J. Am. Chem. Soc. 82, 4427.
E. S. Shubina, E. V. Bakhmutova, A. M. Filin, I. B. Sivaev, L. N. Teplitskaya, A. L. Chistyakov, I. V. Stankevich, V. I. Bakhmutov, V. I. Bregadze, and L. M. Epstein (2002). J. Organomet. Chem. 657, 155.
K. Izod, W. McFarlane, B. V. Tyson, I. Carr, W. Clegg, and W. R. Harrington (2006). Organometallics 25, 1135.
K. Izod, C. Wills, W. Clegg, and R. W. Harrington (2009). Organometallics 28, 2211.
J. Kim, S. Kim, and Y. Do (1992). J. Chem. Soc. Chem. Commun. 13, 938.
K. Izod, C. Wills, M. R. Probert, and R. W. Harrington (2014). Main Group Met. Chem. 37, 113.
K. Izod, C. M. Dixon, R. W. Harrington, and M. R. Probert (2015). Chem. Commun. 51, 679.
D. A. Saulys, N. A. Kutz, and J. A. Morrison (1983). Inorg. Chem. 22, 1821.
K. Vaas, Doctoral Thesis (University of Stuttgart, Stuttgart, 1999).
S. I. Uspenskaya, K. A. Solntsev, and N. T. Kuznetsov (1975). Zh. Strukt. Khim. 16, 482.
I. Tiritiris, Th. Schleid, K. Müller, and W. Preetz (2000). Z. Anorg. Allg. Chem. 626, 323.
Z. Otwinowski and W. Minor, Scalepack: Program for Empirical Absorption Correction (University of Texas at Dallas, Richardson, 1997).
G. M. Sheldrick, SHELX: Program System for Crystal Structure Determination, Solution and Refinement from Diffractometer Data (University of Göttingen, Göttingen, 1997).
R. X. Fischer and E. Tillmanns (1988). Acta Crystallogr. C 44, 775.
Acknowledgements
We thank Dr. Falk Lissner and Dr. Sabine Strobel for the collection of the single-crystal X-ray diffractometer data. Furthermore, we like to thank Dr. Florian Ledderboge and Dipl.-Chem. Adrian H. Geyer for the single-crystal Raman measurements. We are also grateful to Dr. Katharina Bader from the group of Prof. Dr. Joris van Slageren (Institute for Physical Chemistry, University of Stuttgart) for the practical instruction to FT/IR measurements using potassium-bromide pellets.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kleeberg, F.M., Zimmermann, L.W. & Schleid, T. The Crystal Structures of Two Hydro-closo-Borates with Divalent Tin in Comparison: Sn(H2O)3[B10H10] · 3 H2O and Sn(H2O)3[B12H12] · 4 H2O. J Clust Sci 33, 2489–2497 (2022). https://doi.org/10.1007/s10876-021-02166-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-021-02166-6