
Abstract
Geochemical effects of electro-osmosis in bentonite clay are studied in the laboratory, where a 6 mm thick bentonite layer is subjected to direct current. Acidification and alkalization near anode and cathode are expected, possibly causing mineral deterioration, ion mobilization and precipitation of new solids. Afterwards the clay is analysed by XRF and anolyte and catholyte are analysed by ICP-MS. In addition, as a preliminary experiment treated bentonite is analysed by high resolution μ-XRF. Electro-osmotic flow is observed. Due to its carbonate content the bentonite is pH-buffering. Alkalization in the catholyte is substantial. Ca, Na and Sr are significantly removed from the clay and accumulate in the catholyte. Recovery in the catholyte accounts for a small fraction of the element-loss from the clay. The rest will have precipitated in undetected solid phases. μ-XRF indicates the loss of Ca-content throughout the bentonite layer.
Similar content being viewed by others


1 Introduction
Electro-osmosis in clayey soils and sediments, for purposes of clay consolidation, dewatering or cleanup often fails because of unknown geochemical side effects [1]. Electro-osmosis has side effects due to electrolysis of water at large voltages, causing production of H+ at the anode and of OH− at the cathode. In clays of poor acid buffering capacity a low-pH front moves through the clay towards the cathode during electro-osmosis. If the clay does contain carbonates, the pH at the anode will not drop below about 7. However, at the cathode pH may rise to a high level. Extremely low and high pH leads to deterioration of the clay crystal or surface composition; e.g. iron, aluminium, sodium and/or calcium may be mobilized. They subsequently may precipitate as salts or hydroxides, leading to declining electro-osmotic flow. In addition the surface charge of the clay minerals may change.
The aim of the current study is to observe possible changes in the chemical composition of a bentonite clay during electro-osmosis in the laboratory. Changes are studied by XRF-measurements in the wet bulk clay, mainly determined by the solid phase composition, and by pH and ICP-MS measurements in the influent and effluent solutions of the clay. Moreover, electro-osmotic flow of the pore solution is recorded.
2 Literature
The electro-osmotic volume flux of a solution (J v in m s−1) is expressed by the following phenomenological equation:
where E is the electrical potential (V), k e is the coefficient of electro-osmotic conductivity (m2 V−1 s−1).
Bolt [2] explains how electro-osmotic conductivity of clays can be predicted from the Gouy-Chapman diffuse double layer theory. According to Mitchell [3] the practical range for k e of fine-grained soils is between 1 × 10−5 and 1 × 10−4 cm2 V−1 s−1.
Several studies on electro-osmosis have revealed geochemical side effects. Grundl and Michalski [4] and Chen and Murdoch [5] discuss the effect of electrolysis of water that occurs at large voltages, causing production of H+ and O2 (g) at the anode and of OH− and H2 (g) at the cathode. In carbonate-deficient soils the resulting low pH front moves from the anode through the clay meeting the hydroxyl-ions near the cathode. Since at the low pH soil–Fe and –Al and contaminant heavy metals become mobile, they may precipitate as salts or (hydr)oxides near the cathode, leading to declining electro-osmotic and hydraulic conductivities [6]. The formation of precipitates of Fe- and Al-hydroxides and carbonates adds to the electro-osmotic stabilization of clay soil [7, 8]. If a clayey soil does contain lime, the proton production at the anode is buffered in the soil and pH does not drop below about 7. However, at the cathode pH will rise to a high range of 10–11 [4], both in the presence and absence of lime.
Geochemical changes in the clay are expected to lead to a change in the electro-osmotic conductivity, and therefore in the electro-osmotic water flow rate [1, 6]. Causes for these effects are changing surface charge of the clay minerals (depending on their pH-dependency), changing cation occupation at the exchange complex and/or precipitates formed in the pores. In addition low pH will lead to deterioration of the clay crystal structure.
3 Materials and methods
3.1 The clay material
A Wyoming Na-bentonite, (Colclay A90® Ankerpoort, Geertruidenberg, The Netherlands) was used in the experiments. The initial chemical composition of the bentonite is presented in Table 2 (to be discussed later) as total element contents measured by XRF. Table 1 presents physico-chemical properties, adopted from Keijzer and Loch [9]. However, the carbonate content is estimated separately from the XRF-analyses of Ca and Mg, taking the fractions of these cations on the exchange complex (Table 1) into account.
3.2 Experimental methods
An experimental cell was designed for the study of electro-osmotic flow and geochemical side effects in the bentonite clay at a constant current density of 0.005 A m−2 (see Fig. 1). The cylindrical cell, made of PVC consists of a top and bottom part in between which a layer of 10 g of dry bentonite is fit in a circular space of about 6 mm thick and 5 cm diameter. The resulting bulk density of the clay is 0.85 g cm−3. On each side of this clay slab a porous stone is present of about 5 mm thick. On the interface of each porous stone with the clay a gold wire is placed to function as electrodes for current supply. By means of tygon tubing through top and bottom of the cell the clay is in contact with equilibrium reservoirs containing influent and effluent aqueous solutions. Initial equilibrium composition of both reservoirs is 10 mmol L−1 NaCl in demineralized water. Water saturated clay is placed in the cell and subsequently brought in equilibrium with the reservoir solutions during 5 days. No additives are applied to the reservoirs. Each reservoir is connected to a capillary standpipe with an internal diameter of 2.93 mm. Thus, inflow and outflow rates are monitored. The volumes of anolyte and catholyte reservoirs are about 10 mL each. The volume of the water saturated bentonite is kept constant by means of clamp screws, inserted in the cell, to prevent it from swelling. Throughout the experiments both reservoirs are continuously recirculated.

Experimental cell
During the electro-osmotic experiment the pH is monitored in both the anolyte and catholyte reservoirs. Furthermore, the voltage difference between the gold electrodes is monitored as well as the reservoir voltages. Reservoir voltages are monitored by Ag/AgCl-electrodes, indicated as reference electrodes in Fig. 1. Since the salt concentration in the reservoirs are kept homogeneous and in equilibrium with the pore water in the clay, the voltage drop over the reference electrodes represents the real voltage drop over the clay slab, excluding polarization potentials at the gold electrodes. Both electro-osmotic flow and pH are recorded for 34 days. After termination of electro-osmosis the clay layer is cut in four slices perpendicular to the flow direction. Each slice is analysed again by XRF.
The concentration of ions in the anolyte and catholyte solutions is measured at the end of the experiment by ICP-MS analyses. XRF- and ICP-measurements are aimed at observing possible chemical changes in the clay.
A benchtop μ-XRF system (Edax Orbis®, X-ray optics with capillary focussing to 30 μm spotsize) is applied on a cross section of the bentonite slab along the flow path before and after electro-osmosis, to obtain a high resolution scan of the chemical composition of the material. μ-XRF is expected to give indications of changes in the chemical structure of the clay.
4 Results and discussion
Whereas a fixed current density was applied, the resulting voltage varied strongly in time as can be seen in Fig. 2. In the first 100 h a voltage drop along the sample of 100 mV was observed, resulting in an overall gradient of 16.7 V m−1. After about 350 h a voltage drop of 0.01 V resulted in an overall gradient of 1.7 V m−1. For the first 100 h an electrical conductivity of 0.31 mS m−1 is derived. After 300 h the electrical conductivity increased a factor 10 to 3.1 mS m−1. These values are below the range of electrical conductivities for fine grained soils, reported by Mitchell [3] and Heister et al. [10], who report a minimum value of 10 mS m−1.

Voltage drop along the clay sample at a constant current density of 0.005 A m−2
In Fig. 3 the water flow into/out of the clay at the anode and at the cathode side is presented. During the first 240 h of current application both standpipes showed intake of water into the bentonite. After that the flow in the catholyte standpipe reversed and outflow of solution from the clay, simultaneously with inflow at the anolyte standpipe, got going. From Fig. 3 it can be derived that, due to water behaviour during the first 240 h, there is a net accumulation of 0.85 cm3 water in the clay which must have coincided with cell expansion. The cause of this water behaviour is unknown. No gas formation was observed at the gold electrodes. This is confirmed by the inward direction of the water fluxes during the first 240 h. By linearization of the flow in the cathode standpipe between 300 and 800 h and making use of the voltage gradient derived from Fig. 2, a coefficient of electro-osmotic conductivity (Eq. 1) for the bentonite is derived:
Although between 250 and 300 h the flux to the catholyte was larger, one derives from Figs. 2 and 3 that k e remains nearly the same. Figure 3 shows that beyond 600 h the water flux is diminishing, which, at the constant voltage in that range, implies a k e decreasing below the above value. Compared with values reported elsewhere, e.g. 1 × 10−5 cm2 V−1 s−1 in Boston blue clay by Alshawabkeh et al. [11], 1 × 10−6 to 8 × 10−7 cm2 V−1 s−1 in bentonite by Heister et al. [10] and the values reported by Mitchell [3], this coefficient is at the low end of theses ranges. This may be explained by the buildup of a hydraulic head difference in the standpipes up to 17 cm.

Electro-osmotic flow in bentonite; blocks represent the catholyte standpipe and diamonds the anolyte standpipe. Initial positions of the standpipe levels are taken zero
The development of pH in anolyte and catholyte is shown in Fig. 4. pH in the anolyte stays constant at about the initial value of 7.5. The carbonate-content of the bentonite provides a sufficiently large proton buffer to keep pH in the anolyte constant at the pH of the bentonite. At the end of the experiment the sum of the cations Ca and Mg is still far exceeding the CEC (as can be derived from Table 2, presented hereafter). Therefore, acid-weathering of the clay is not expected. In the catholyte the pH stays at this value during the first 25 days, after which it rises and varies between 8 and >14. Thus, clay mineral deterioration can only occur by alkalization, possibly mobilizing macro-elements from the crystal grid.

pH-changes in anolyte (+) an catholyte (−) during electro-osmosis in bentonite. During the first 10 days no electro-osmotic flow was observed
Element analysis by XRF was done on 2 untreated bentonite samples (initial contents) and again on 4 bentonite slices cut after electro-osmotic treatment (final contents). The results are presented in Table 2. The standard deviations in Table 2 are sufficiently low to allow a good comparison between initial and final element contents. Thus, significant decreases are observed for Fe, Mn, Ca, Na, Ba and Sr. For Al a slight increase is observed.
Of the initial Ca-content of 1.92% in the clay, about 0.48% is estimated to occupy the CEC. The rest is assumed to be in the form of CaCO3. Since the Mg-occupation of the CEC is very low (Table 1), the initial Mg-content is assumed to be all in the form of MgCO3. The initial Na-content amounts to about 85% Na+-occupation of the CEC and has decreased to 35% of the CEC at the end of the experiment. Na+ on the CEC must have been replaced by Ca2+ and Mg2+, dissolved from their carbonates by proton input from the anolyte, which are still significantly present at the end of the experiment. Since Sr-behaviour in soils is mainly governed by easily soluble SrCO3-precipitates and Ca/Sr-ion exchange [12], the initial Sr in the bentonite can be both in carbonate form and on the exchange complex. Since Ba has the tendency to form salts with limited solubility in soil and water, its initial presence in the clay is expected to be on the exchange complex. Both Sr2+ and Ba2+ are partially lost by cation exchange with Ca2+. The loss of Fe and Mn is supposedly by reduction.
In Table 3 final cation concentrations in anolyte and catholyte are presented. It is noted that, whereas initially only Na+ is present as cation in the reservoirs, at 10 mmol L−1, the anolyte has slightly increased in Ca, Mg and S by probable diffusion from the bentonite. However, the catholyte concentrations show larger increases in Ca2+, Mg2+, Na+, Sr2+, S and Si. The concentrations of Al, Fe, Mn and Ba in the catholyte are <5 μmol/kg. Their loss from the clay is not recovered in the catholyte solution. For Fe and Mn this may be explained by formation of insoluble (hydr)oxides, for Ba by formation of insoluble salts in the catholyte reservoir.
The loss of Ca, Na, Sr and Si from the clay coincides with relatively large excess concentrations in the catholyte, although their quantities in the catholyte account for only resp. 10, 28, 13 and 1% of their loss from the clay. Mass balance calculations using Tables 1, 2 and 3 show that 43% of the initial Ca-content is recovered in clay and reservoirs at the end of the experiment. For Na, Sr and Si this is resp. 60, 64 and 99%. Si and S in the catholyte may have been transported from the clay in complexed forms with cations. The increase of Mg in the catholyte is not consistent with its increasing content in the clay. The overall low recovery of elements suggests that some loss occurs during the experiment. A probable explanation for the unaccounted quantities of Ca and Sr is their reprecipitation as carbonate in the catholyte reservoir and in the porous stone.
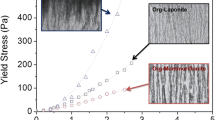
As a preliminary study μ-XRF has been applied to the cross section of the sample along the flow path. The results of two scans, representing untreated and treated material are presented in Fig. 5.

Distribution of macro-elements along the flow path in bentonite before and after exposure to electro-osmosis, as observed by μ-XRF
According to these scans the relative element contents appear to be reduced, for Ca by a factor of about 2 (see Table 2 and Fig. 5). The element contents after treatment appear to be more homogeneously distributed in the clay than before treatment. This may indicate that mobilization of the elements by electro-migration is completed by the end of the experiment, at the relatively low current applied.
Since only for Ca, Na and Sr the concentration changes in both clay and catholyte reservoir are significant and consistent, some interpretations of their behaviour can be made. Although the pH at the anode side of the clay does not drop below 7.5, proton production does occur but is buffered by dissolution of the carbonates in the clay. So electro-migration of Ca2+ from dissolved CaCO3 is expected, but subsequent Ca/Na-cation exchange on the CEC and precipitation (segregation) as a solid Ca-phase takes place in the clay. Ca-removal from the bentonite is also observed in the μ-XRF scans. In addition the electrical field mobilizes the exchangeably adsorbed (double layer) and free solution Ca2+ ions towards the catholyte and precipitation sites in the clay. Electro-migration of adsorbed and free solution Na+ ions is also the cause of Na+ accumulation in the catholyte reservoir. The release of Sr2+ from the clay originates from carbonate dissolution and from the exchange complex. The Si- and Al-data indicate that clay mineral deterioration has been minor or absent. Therefore, and because of low pH dependence of the surface charge of bentonite, also a change in surface charge will be minor or absent.
5 Conclusions
The conclusions to be drawn from this study are the following. Electro-osmotic flow is demonstrated in bentonite clay, however, the coefficient of electro-osmotic conductivity is lower than reported elsewhere. The bentonite is pH-buffering by its high carbonate content, preventing acid weathering of the clay. Alkalization in the catholyte is substantial. The study does not give sufficient evidence that this has led to silicate mineral weathering. Reduced electro-osmotic flow towards the end of the experiment may be explained by the development of a hydraulic head difference counteracting electro-osmosis. Ca, Na and Sr are significantly removed from the clay and accumulate in the catholyte solution. Ca and Sr (and Mg) mobilization started by carbonate dissolution and subsequent electro-osmotic convection. The Na accumulation in the catholyte originates from the diffuse double layer. Recovery in the catholyte accounts for a small fraction of the element-loss from the clay. The rest will have precipitated in an undetected solid phase.
Preliminary μ-XRF analyses on bentonite indicate a redistribution of elements along the studied profile. XRF and μ-XRF analyses are consistent, where μ-XRF gives a greater detail and potential trends can be spotted.
References
Saichek RE, Reddy KR (2005) Crit Rev Environ Technol 35(2):115–192
Bolt GH (1979) In: Bolt GH (ed) Soil chemistry. B. Physico-chemical models. Elsevier, Amsterdam
Mitchell JK (1993) Fundamentals of soil behavior, 2nd edn. Wiley, New York
Grundl T, Michalski P (1996) Water Res 30:811–818
Chen J-L, Murdoch L (1999) J Geotech Geoenviron Eng 125:1090–1098
Cherepy NJ, Wildenschild D (2003) Environ Sci Technol 37:3024–3030
Bjerrum L, Moum J, Eide O (1967) Géotechnique 17:214–235
Azzam R, Oey W (2001) Transp Porous Media 42:293–314
Keijzer TJS, Loch JPG (2001) Soil Sci Soc Am J 65:1045–1055
Heister K, Kleingeld PJ, Loch JPG (2005) J Colloid Interface Sci 286:294–302
Alshawabkeh AN, Sheahan TC, Wu X (2004) Mech Mater 36:453–465
Lefevre F, Sardin M, Schweich D (1993) J Contam Hydrol 13:215–229
Acknowledgements
This publication was based on work supported by Award No KUK-C1-017-12, made by King Abdullah University of Science and Technology (KAUST).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Loch, J.P.G., Lima, A.T. & Kleingeld, P.J. Geochemical effects of electro-osmosis in clays. J Appl Electrochem 40, 1249–1254 (2010). https://doi.org/10.1007/s10800-010-0098-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-010-0098-6