
Abstract
Clonal and genetic diversity in foundational plant species are critical for species resiliency and ecosystem processes, both of which contribute to restoration success; however, genetic data is often lacking for common plant species used in many restoration practices. Only a few plant species dominate salt marshes, ecologically and economically valuable ecosystems targeted for restoration due to global decline. Despite being a foundational species in southeastern United States salt marshes black needlerush (Juncus roemerianus Scheele) is understudied, especially in comparison to the co-occurring smooth cordgrass (Spartina alterniflora Loisel). We used a panel of 18 microsatellite markers on 849 samples of J. roemerianus collected at 17 sites across a majority of the species range from Mississippi to South Carolina to measure clonal and genetic diversity and characterize population structure. Results are consistent with previous genetic studies on J. roemerianus and other clonal plant species in that clonal and genetic diversity were higher than expected based on life history literature, with an average genotypic diversity (GD) of 0.67 and average observed heterozygosity (HO) of 0.56. Differences in diversity between the ecologically and environmentally divergent Gulf (GD = 0.64, HO = 0.52) and Atlantic (GD = 0.80, HO = 0.45) coasts suggest that life history strategy may vary by environment. Hierarchical structure was apparent across the study area, with STRUCTURE analyses identifying three genetic clusters that further subdivided into five clusters and a broad transition zone. The scale of this genetic differentiation should be considered in sourcing plants for salt marsh restoration efforts.
Similar content being viewed by others

Introduction
Common, foundational plant species are key components of ecological restoration that seeks to reestablish ecosystem processes and benefits (Hughes et al. 2008; Reusch and Hughes 2006), but have been understudied in conservation genetics (McKay et al. 2005). Native plant restoration success is dependent on the genetic composition of source material (Hufford and Mazer 2003; Mijnsbrugge et al. 2010), specifically the amount of genetic diversity and genetic similarity to the restored area, which can be estimated using neutral genetic markers (McKay et al. 2005). Genetic diversity of dominant plant species in monotypic landscapes is analogous to species diversity in maintaining ecosystem processes (Hughes et al. 2008; Reusch and Hughes 2006), and contributes to restoration success (Reynolds et al. 2012). In clonal plant species, preserving natural levels of clonal diversity (number of clonal variants) improves plant growth and reproduction (Williams 2001; Hämmerli and Reusch 2003; Reusch et al. 2005), and increases ecosystem services (Hughes and Stachowicz 2009; Crutsinger et al. 2006). Salt marshes are monotypic landscapes dominated by a few clonal plant species (Stout 1984) that are undergoing restoration nationally and globally, but have been generally understudied in the conservation genetics literature.
Decades of destruction and degradation by waste discharge and land conversion (Broome et al. 1988) has caused salt marshes to decline by 13–30% worldwide with an over 50% decline in the United States (Kennish 2001; Valiela et al. 2009). These habitats are further threatened by climate change and sea level rise (Gedan et al. 2009). More recently, the value of salt marshes to human health and wealth, as well as wildlife, has prompted legislative protection and restoration efforts. Salt marshes supply ecosystem services valued at $10,000 per hectare (Kirwan and Megonigal 2013; Zedler and Kercher 2005), and provide critical habitat to a number of ecologically and economically important species (Kennish 2001). The southern Atlantic coast of the United States, from Florida to North Carolina, and Gulf of Mexico coasts of the United States harbor a significant portion of the remaining salt marsh habitat in the world, supporting 12,440 km2 of tidal marsh that makes up 78% of the United States tidal marsh area and 27% of global marsh area (Greenberg and Maldonado 2006; Greenberg et al. 2006). In the southeastern United States, the two primary target plant species for restoration are smooth cordgrass (Spartina alterniflora Loisel) and black needlerush (Juncus roemerianus Scheele). Although clonal and population genetic analyses have been conducted on S. alterniflora (ex. Novy et al. 2010; Richards et al. 2004), to our knowledge, only a single study has been conducted on J. roemerianus (Tumas et al. 2018).
Juncus roemerianus is a clonal, gynodioecious macrophyte and a foundation species in salt marshes from eastern Texas to the Mid-Atlantic in Maryland (Godfrey and Wooten 1979; Eleuterius 1976; Stout 1984). The species creates marsh area by accreting and stabilizing sediment, providing habitat for other salt marsh wildlife (Pennings and Bertness 2001). Life history literature on the species reports J. roemerianus as using primarily clonal reproduction with sexual reproduction only occurring during rare recruitment events to colonize new areas (Stout 1984), implying that populations of J. roemerianus are composed of only a few clonal variants. Genetic data are needed to inform restoration ecologists about the rate of clonal reproduction, and how rates vary across environments in natural populations, so that restored ecosystems reflect natural levels of resiliency and diversity.
The only genetic study on J. roemerianus to date examined clonal and genetic diversity across ten sites in the northeastern Gulf of Mexico from eastern Mississippi to the western panhandle of Florida (Tumas et al. 2018). As reported for other clonal plant species (Ellstrand and Roose 1987; Silvertown 2008; Gabrielsen and Brochmann 1998; Lloyd et al. 2011; Pleuss and Stocklin 2004), clonal and genetic diversity were higher than expected based on the existing life history literature with an average genotypic diversity of 0.54 and average observed heterozygosity of 0.58 across the ten sites. However, the study only examined a portion of the geographic range of J. roemerianus and did not include the environmentally and ecologically divergent Atlantic coast (Dardeau et al. 1992). Genetic patterns need to be examined on a broad scale to adequately inform restoration and management across the species range, and comparing genetic results from different environments may reveal additional information on J. roemerianus biology and life history.
We performed a broad-scale population genetic analysis on J. roemerianus across the majority of the species range in the southeastern United States from western Mississippi to South Carolina to expand existing studies and inform tidal marsh restoration practices. The objectives of our study were to (i) measure standing levels of clonal and genetic diversity in natural populations, (ii) compare genetic patterns between the Atlantic and Gulf coasts, and (iii) delineate broad-scale population structure across the majority of the species range. Based on the recent genetic study on J. roemerianus, we hypothesized clonal and genetic diversity would be high and similar across the study area, and that the strongest genetic differentiation would occur between the Atlantic and Gulf coasts. Results from the study will provide valuable information for the conservation and restoration of critical coastal ecosystems.
Materials and methods
Sample collection
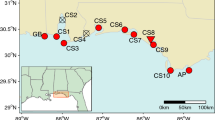
Seventeen sites were sampled across the northeastern Gulf of Mexico and southeastern Atlantic coast from Moss Point, MS to Awendaw, SC (Fig. 1). Ten sites were sampled in the Gulf of Mexico as part of another study (Tumas et al. 2018), two of which (GB and AP) are National Estuarine Research Reserves (NERR). A set of 304 samples was collected from Grand Bay NERR (GB) in January and March 2015, and a set of 32 samples was collected from Apalachicola NERR (AP) in May 2015 and March 2016. Thirty samples were collected from eight sites (CS1, CS3, CS5, CS6, CS7, CS8, CS9, CS10) between the two NERRs in March 2016. Forty samples were collected randomly from an additional seven sites in November 2016 for this study to extend the range in the Gulf coast (EC1 and EC2), and include the Atlantic coast (EC3, EC4, EC6, EC7, EC8) (Table 1). Samples consisted of a single leaf collected and stored in plastic bags, and a GPS waypoint taken using a Garmin GPSMAP 64st. Distances among sites ranged from 14 to 875 km with an average of 366 km. Distance among samples within sites ranged from 0.1 m at CS8 to 14 km at AP with an average of 4 km, varying based on area of marsh at each site.

Maps showing allelic richness (top) and genotypic diversity (bottom) across the 17 sites on the Atlantic and Gulf coast. Atlantic coast sites have overall greater genotypic diversity and a greater proportion of multilocus genotypes (MLG, blue) to clonal replicates (C, red) compared to Gulf coast sites, while Gulf coast sites have greater genetic diversity. Site labels are in black in the top map. (Color figure online)
Microsatellite amplification
Nineteen species-specific microsatellite markers (Tumas et al. 2017) were used to genotype samples following DNA extraction protocol, PCR reaction conditions, and thermal cycling parameters described in Tumas et al. (2017). Samples were successfully scored across a minimum of 15 loci, or genotyping was repeated on the sample. Genotyping error rate was calculated by re-genotyping approximately 17% of samples and dividing the number of mismatched loci identified by Identity Analysis in CERVUS (Kalinowski et al. 2007) by the total number of scored loci.
Clonal analysis
Clonal analyses are sensitive to missing data, which can lead to an overestimation of unique genotypes. Therefore, a subset of twelve markers (Jr03, Jr05, Jr12, Jr13, Jr16, Jr33, Jr41, Jr42, Jr46, Jr58, Jr80, Jr86) that had the least missing data were selected and only samples with complete 12 locus genotypes were used in clonal analyses. The selected markers have a combined probability of exclusion of 1.03 × 10−10. Unique multilocus genotypes (MLG) were identified, and clonal metrics were calculated using the package poppr v2.5 (Kamvar et al. 2014, 2015; R Core Team 2016). The metrics psex and pgen were calculated across each sample using the ‘psex’ and ‘pgen’ functions respectively. Number of MLGs, Simpson’s index corrected for sample size (λ), Shannon–Wiener Index of MLG diversity (H), genotypic diversity (GD), and genotypic evenness (E.5) (Grünwald et al. 2003) were calculated across samples grouped by site and coast of origin (Gulf of Mexico or Atlantic). Genotypic diversity, or the number of unique genotypes, was not reported in poppr and was calculated as (G − 1)/(N − 1) where G is the number of unique MLGs at a site and N is the total number of ramets sampled (Arnaud-Haond et al. 2007). Simpson’s index was corrected for sample size by multiplying the reported lambda by (N/(N − 1)) where N is the total number of ramets sampled.
Genetic diversity analyses
The full panel of markers was used in genetic diversity and structure analyses, following removal of genetically identical clonal replicates identified using Identity Analysis in CERVUS. A single ramet was randomly selected from each group of genetically identical samples. Hardy–Weinberg Equilibrium was tested at each locus across samples within each site using CERVUS. Linkage disequilibrium was calculated between pairs of loci at each site using Genepop (Raymond and Rousset 1995).
Expected (HE) and observed (HO) heterozygosity, allelic diversity (AD), and the inbreeding coefficient (FIS) were measured across sites using Arlequin (Excoffier and Lischer 2010). Allelic richness (AR) within sites rarefied for sample size was calculated using the divBasic function in the ‘diveRsity’ package in R (Keenan et al. 2013). The number of private alleles was measured using the private_alleles function in poppr. Genetic diversity metrics were also calculated across samples grouped by genetic cluster and coast of origin.
Statistical analysis
Average and minimum distance among samples within a site were correlated to measures of clonal diversity, and site area was correlated to clonal and genetic diversity to ensure the sampling methods did not arbitrarily affect results. Correlations were conducted in R using the Pearson’s correlation method and only clonal (GD, λ, E.5) and genetic (AR, HO, HE, FIS) diversity metrics that accounted for samples size were used in this analysis. A Student’s two-sample t-test was performed on site specific values of GD, HO, and AR categorized by coast in R to test for significant differences between the Atlantic and Gulf coast. Statistical significance was based on an alpha value of 0.05 for all analyses.
Canonical correspondence analysis (CCA) was performed using the vegan package (Oksanen et al. 2019) in R to examine the influence of the surrounding landscape on genetic diversity. CCA has been successfully used to identify landscape variables that significantly contribute to variation in genetic diversity (Angers et al. 2002; Cleary et al. 2017). Two models were run using two different genetic diversity metrics (HO and AR) as the dependent variable, represented as matrices of locus-specific values per site. Missing data was removed from the genetic diversity matrices, resulting in the loss of one site (CS8) and three loci (Jr05, Jr72, Jr80). Explanatory variables were represented by four landscape variables: proportion wetland, proportion developed land, tide level, and distance to high intensity developed land. Proportion wetland in the surrounding landscape could increase genetic diversity through gene flow from other J. roemerianus populations, while proportion of developed land or close proximity urban areas may decrease or disrupt gene flow (Tumas et al. 2018). Flooding amplitude and frequency, represented by tidal level, influences many environmental and ecological characteristics in the salt marsh including plant community composition (Pennings and Bertness 2001), and are a primary difference between the Gulf and Atlantic coasts (Dardeau et al. 1992).
Land cover data was measured using the National Oceanic and Atmospheric Administration’s coastal change analysis program (C-CAP) land cover atlas for Mississippi, Alabama, Florida, Georgia, and South Carolina (Coastal change analysis program (C-CAP) 2015/2016 Regional Land Cover Data—Contiguous United States, Department of Commerce, NOAA, National Ocean Service, Office for Coastal Management). Three raster layers were created in ArcGIS v10.4.1 to represent wetland, defined as estuarine or palustrine categories; developed land, defined as open, low, medium, and high intensity developed land categories; and high intensity developed land, defined as 80–100% constructed materials (Table S1). Layers were created by reclassifying the land cover categories of interest to one and all other categories to zero. The high intensity developed land layer was converted to a polygon and distance to high intensity developed land was calculated using the “Join and Relates” function in ArcGIS. Proportion of wetland and developed land within a 1 km buffer was calculated in R from the respective raster layers using the raster package (Hijmans 2019). Tide level at each site was determined using NOAA’s Center for Operational Oceanographic Products and Services Tides and Currents dataset, taking “Mean Tide Level” from the nearest station with a water level sensor (Table S2). Variance inflation factors were calculated to assess multicollinearity, allowing a maximum value of two. ANOVA like permutation tests were used to determine the significance of the two models and the significance of each landscape variable. Genetic diversity was plotted against landscape variables identified as significant to assess the directionality of the relationship.
Population structure analyses
Genetic clusters were identified using the program STRUCTURE via 20 iterations of 100,000 steps following a burn-in period of 50,000 with admixture, with and without collection sites used as prior locations, and correlated allele frequencies for each of K = 1–6 (Pritchard et al. 2000). Additional STRUCTURE runs following the same parameters were conducted on samples grouped by genetic cluster to examine hierarchical genetic structure. The optimal number of k clusters was chosen using the Evanno method in STRUCTURE HARVESTER (Earl and vonHoldt 2012). Individual cluster assignment was determined by maximum Q value, and sites were assigned based on the assignment of the majority of resident individuals. Principal components analysis (PCA) on the variance-covariance matrix of allele frequencies was conducted using the dudi.pca function in the ‘ade4’ package in R (Dray and Dufour 2007; Chessel et al. 2004; Dray et al. 2007). PCA examines continuous genetic structure based on genetic similarity, unlike STRUCTURE that groups samples into discrete clusters so that frequencies meet Hardy–Weinberg Equilibrium expectations. Pairwise genetic distances among sites and clusters were measured as Slatkin’s linearized FST (D = FST/1 − FST) using Arlequin, correcting associated p-values for false discovery rate using the Benjamini and Yekutieli method (Narum 2006). A Mantel test was run on the Slatkin’s linearized FST pairwise genetic distances using the mantel.randtest function in the R-package ‘ade4’ to test for significant patterns of isolation by distance (IBD). An IBD plot was generated by plotting Slatkin’s linearized FST against log transformed geographic distances.
Results
Clonal diversity
Across the subset of twelve markers used in clonal analyses, 692 samples had no missing data from which 472 samples were identified as unique MLGs. All MLGs had psex < 5.81 × 10−6 and pgen < 1.23 × 10−8. Clonal diversity and evenness varied across sites with an average Simpson’s Index of 0.92 and average Shannon-Weiner Index of MLG diversity of 2.85. Genotypic evenness ranged from 0.49 at site EC1 to 1.0, with an average genotypic evenness of 0.85. Genotypic diversity ranged from 0.08 at CS8 to 1.0 at two sites (CS10 and EC7) composed entirely of unique MLGs, and with an average of 0.67 (Table 1). Gulf coast samples had a lower genotypic diversity (GD = 0.64) than the Atlantic coast (GD = 0.80) (Fig. 1), but an equal Simpson’s Index and higher Shannon-Wiener Index (λ = 0.996, H = 5.56) than the Atlantic coast (λ = 0.996, H = 4.83). Genotypes were more evenly abundant across Atlantic coast samples (E.5 = 0.84) than across Gulf of Mexico samples (E.5 = 0.67) (Table 1).
Genetic diversity
A total of 849 samples were typed across a minimum of 15 loci in the full panel of microsatellite markers, and 529 samples were identified as unique genotypes for use in genetic diversity and population structure analyses. Overall genotyping error rate was 2.08%. One marker (Jr73) deviated from Hardy–Weinberg Equilibrium at six sites (EC1, EC2, EC3, EC4, EC7, and EC8), and was subsequently removed from all other analyses. Twelve pairs of loci exhibited linkage disequilibrium across five sites and were not excluded from analysis (Table S3).
Genetic diversity was moderate across sites, with an average expected heterozygosity of 0.53, an average observed heterozygosity of 0.56, and average allelic richness of 2.33 alleles per locus (Table 1). The Gulf coast had higher values of genetic diversity (HE = 0.65, HO = 0.52, AR = 6.96) than the Atlantic coast (HE = 0.53, HO = 0.45, AR = 5.26) (Fig. 1). Samples across the Gulf of Mexico also had a greater number of private alleles (32) than samples from the Atlantic coast (4). Levels of inbreeding varied across sites (average FIS = − 0.02), but no site had a significant FIS value following a sequential Bonferroni correction. Gulf coast samples had a slightly higher FIS (0.17) than Atlantic coast samples (0.14).
Statistical analysis
Average and minimum sampling distances were not significantly correlated to the tested measures of clonal diversity, and site area was not significantly correlated to any of the tested metrics of clonal or genetic diversity (p > 0.05; Table S4). Results of the t-test showed GD (p = 0.01) and HO (p = 0.02) were significantly different between Atlantic and Gulf coasts, but AR (p = 0.47) did not significantly vary between the two coasts. The CCA demonstrated that landscape variables explain a significant portion of the variation in HO (p = 0.04) among sites, but not AR (p = 0.32) based on model-level ANOVA like permutation tests. The four landscape variables explained 37% of the variation in HO, with the first and second canonical axes explaining 54% and 34% of the constrained variation, respectively. The only significant landscape variable was tide level (p = 0.02), which had a negative relationship with HO (Figure S1).
Population structure
The optimal number of genetic clusters from the STRUCTURE analyses was k = 2 when collection sites were not considered as priors, with sites CS8–EC3 forming an eastern Gulf Coast cluster (EGC) and all remaining sites in the western Gulf and Atlantic forming a second cluster (Fig. 2). When collection sites were set as prior locations, k = 3 was the optimal clustering. The second cluster above was subdivided into a western Gulf Coast (WGC) cluster (GB–CS7) and a South Atlantic Bight (SAB) cluster (EC4–EC8). The southernmost site on the Atlantic Coast (EC3) grouped with the ECG cluster. However, some individuals in sites CS5–CS7 were assigned to the EGC cluster and some individuals in sites EC4–EC6 were assigned to either WGC or EGC based on maximum Q value, indicating admixture at these sites. Hierarchical STRUCTURE analyses indicated an optimal number of k = 2 within each of the three genetic clusters, with the two sets of admixed sites forming unique clusters and the peninsular Florida sites (EC1–EC3) clustering together (Fig. 2). Site clustering from the PCA analysis across all samples was congruent with the k = 3 and hierarchical STRUCTURE results, and indicated that SAB is more closely related to WGC than EGC, as suggested by the k = 2 result (Fig. 3; Table S5). Of particular note in the PCA is the highly admixed sites CS5–CS7, which fall between the two tighter groupings of sites GB–CS3 and CS9–AP, and CS7 is closer to CS9–AP. CS8 falls further from CS9–AP, which may be due to the smaller sample size at that site. Furthermore, the sub-cluster EC1–EC3 clustered strongly and separately from the other sites. Although these sites did form a cluster in the hierarchical STRUCTURE analysis, this strong differentiation was not apparent in the STRUCTURE results for the overall dataset. The PCA showed similar hierarchical clustering with sites in each sub-cluster falling close together on the plot (Fig. 3). All pairwise measures of FST were significant following correction for false discovery rate (Table S6). Patterns of genetic similarity from the PCA were reflected in measures of genetic distance, which were smaller between SAB and WGC than between either of these clusters and EGC (Table 2). The Mantel test for isolation by distance was significant (0.55, p = 0.001), indicating that genetic distance increases with geographic distance as shown in the IBD plot (Fig. 4).

Structure plots showing the full set without collection sites set as prior locations when k = 2 (a) and with collection site set as prior locations when k = 3 (b). Hierarchical structure within the three clusters shown in b indicated k = 2 with and without collection site included as prior locations for each cluster: Western Gulf coast (c), Eastern Gulf coast (d), and Southern Atlantic Bight (e)

Sites are plotted based on the first and second axis of a principal component analysis (PCA). Text color corresponds to the cluster each site was assigned in the structure analysis, with black representing the Western Gulf Coast, blue representing the Eastern Gulf Coast, and red representing the Southern Atlantic Bight. (Color figure online)

Isolation by distance (IBD) plot showing the significant relationship from the Mantel test. Genetic distance (Slatkin’s linearized FST) is plotted against log transformed pairwise geographic distance (km). A regression line (red) was added to illustrate the relationship. (Color figure online)
Discussion
Clonal and genetic diversity across sites and coasts
Clonal and genetic diversity of the sample sites were high, consistent with previous results examining a smaller portion of the species range (Tumas et al. 2018). Overall, sites had multiple unique MLGs, with two sites consisting of entirely unique genotypes, and all but a single clone was restricted to a single site. The only clone shared between sites consisted of two samples that were identical across 18 markers, and was shared between sites CS3 and CS5 that are approximately 89 km apart. Heterozygosity was moderate across sites, and many sites had private alleles despite low allelic richness. These genetic patterns are consistent with genetic studies on other clonal plant species, which have detected higher than expected levels of clonal diversity and few widespread clones (Ellstrand and Roose 1987; Widén et al. 1994). The co-occurring salt marsh plant species S. alterniflora, also initially thought to be primarily clonal, has similar clonal diversity to J. roemerianus, with an average Simpson’s diversity of 0.998 across seven sites on Sapelo Island, GA (Richards et al. 2004).
Genetic studies on clonal plants have contradicted expectations of low genetic diversity (Gabrielsen and Brochmann 1998; Lloyd et al. 2011), and J. roemerianus exhibits higher levels of genetic diversity than would be expected from a species with rare sexual reproduction. Populations of S. alterniflora have also been found to have higher than expected genetic diversity, with average observed heterozygosity ranging from 0.6 to 0.73 across 12 loci in a study of New York populations (Novy et al. 2010). In contrast, populations of Spartina maritima across the northern limit of the species range in France were found to be genetically depauperate and formed from only two unique genotypes. These marginal populations of S. maritima have no observed seed production and reproduce almost exclusively through clonal propagation (Yannic et al. 2004). Mature plants of J. roemerianus produce up to 60 seeds per flower that are viable for more than a year (Eleuterius 1975). Higher levels of clonal and genetic diversity, and an almost even ratio of unique genotypes to clonal replicates, suggest a more even balance between clonal propagation and sexual reproduction in J. roemerianus.
This balance appears to shift across the Atlantic and Gulf coasts, as indicated by the significant differences in genotypic diversity and observed heterozygosity. Sites in the Gulf of Mexico have overall greater genetic diversity and lower clonal diversity than sites on the Atlantic coast, which could be the result of differences in plant community and disturbance regime across the two coasts. Differences in genetic diversity may be due to differences in areal coverage of the species on each coast, reflecting the positive relationship that has been documented between genetic diversity and both population size and area (Frankham 1996). J. roemerianus is dominant across the northeastern Gulf of Mexico sample sites (Stout 1984), which means the species covers a greater area and possibly a greater number of microhabitats. On the Atlantic Coast, more frequent flooding and competitive exclusion by S. alterniflora and other species generally restricts J. roemerianus to a smaller area in the upper marsh (Stout 1984). This relationship between genetic diversity and population size and area may also explain the CCA results. The negative relationship between tidal range and observed heterozygosity may be indirectly due to the decrease in J. roemerianus areal coverage with increased tidal flooding. Although the significance of tidal range in the CCA may be spurious altogether, a result of tidal range varying the most between coasts out of the four landscape variables. Alternatively, differences in clonal diversity may be due to differences in disturbance regime following the intermediate disturbance hypothesis (Paine and Vadas 1969; Reusch 2006). The Atlantic coast experiences greater and more frequent disturbance from flooding caused by higher amplitude diurnal tides compared to the irregularly flooded marshes in the northeastern Gulf of Mexico (Dardeau et al. 1992). The subsequent loss of established genets and recruitment of new genets through sexual reproduction caused by an intermediate disturbance regime could allow for the introduction of novel genotypes and enable the Atlantic coast to maintain higher levels of clonal diversity (Eriksson 1993; Reusch 2006).
Differences in clonal and genetic diversity between coasts could also be driven by differences in life history strategies. The Gulf and Atlantic coasts may represent the two ends of the clonal plant life history spectrum that transitions from initial seedling recruitment (ISR) with no subsequent seedling recruitment to repeated seedling recruitment (RSR) throughout a species lifetime (Eriksson 1989). Genetic patterns in Gulf Coast populations resemble an ISR strategy, in which standing genotypic diversity depends on a combination of the number of unique MLGs in the founding population and diversifying selection (Eriksson 1993; Widén et al. 1994). Diversifying selection may be able to act on a small scale in the marsh due to highly variable environmental and ecological factors, which vary on a scale of a meter or less (Pennings and Callaway 2000). Greater availability of microhabitats and area for J. roemerianus in the Gulf of Mexico may also cause clonal propagation to be more important for colonization of stressful environments or uninhabited areas, especially since J. roemerianus has the ability to share resources amongst ramets (Pennings and Callaway 2000). Reduced genotypic evenness across Gulf sites may be an artifact of a few MLGs from the founding population adapting and proliferating in each population, and may further indicate an ISR strategy. Furthermore, populations of S. alterniflora in the Gulf of Mexico experience declines in genotypic diversity with population age following initial establishment, as expected in species that follow an ISR strategy (Travis et al. 2004). Conversely, an intermediate disturbance regime could allow for repeated seedling recruitment via sexual reproduction in Atlantic populations that more closely reflects a RSR strategy, resulting in the observed elevated clonal diversity. While determining life history strategy in J. roemerianus would require a long-term study, environmental factors driving life history strategy are not unique and other species have been demonstrated to use different strategies in different habitats (Eriksson 1993). Furthermore, if the rate of clonal propagation and sexual reproduction is genetically linked, environmental factors could be driving adaptation and leading to functional genetic differences between coasts in J. roemerianus.
Genetic structure
Analyses in STRUCTURE and a PCA revealed that population structure occurs on a relatively broad scale in J. roemerianus. Several sites showed admixture between or among clusters, further indicating that a higher level of sexual reproduction, and therefore gene flow, occurs to create large, admixed populations. Dispersal in J. roemerianus can occur asexually through water transport of vegetative ramets (USDA, NRCS 2018) or sexually through seeds and pollen, which may be transported by water, wind, or animal vector (Cronk and Fennessy 2001). The lack of shared unique genotypes among sites suggests that the observed population structure is a result of sexual reproduction. Many other wind-dispersed plant species also persist in large, effectively panmictic populations even when some population fragments are isolated (Ashley 2010), such as some salt marsh fragments. Water (Huiskes et al. 1995) or bird-mediated (Soons et al. 2008) dispersal would also allow for longer dispersal distances between fragmented salt marsh populations. Admixture in sites CS5–CS7 suggests a cline rather than an abrupt break between the WGC and EGC clusters that could result from the steady decline in gene flow with increasing distance indicated by the IBD plot and significant Mantel test.
Interestingly, genetic similarity among the clusters was not intuitive based on geographic location. The SAB cluster was more closely related to the geographically distant WGC cluster than to the geographically proximal EGC cluster. Substructure within each genetic cluster indicates genetic structure occurs hierarchically in the species, and sub-clusters may indicate the scale at which genetic neighborhoods are formed. While the general pattern of clustering and sub-clustering was shared between the STRUCTURE and PCA results, the strength of some sub-clustering was more pronounced in the PCA. In particular, the three peninsular Florida populations (EC1, EC2, EC3) more strongly clustered out in the PCA. This difference may be due to the difference in underlying theory of the two analyses. While the three sites may show substantial differentiation in allele frequencies, as shown in the PCA and indicated by the high number of private alleles, the three sites may not be in Hardy–Weinberg Equilibrium, causing the degree of differentiation to have been lost in the STRUCTURE results. The conclusions gleaned by examining and comparing both sets of results reiterates the importance of running multiple analyses with different theoretical principles.
The geographic location of the genetic break between genetic clusters of J. roemerianus on the Atlantic coast and Gulf of Mexico, occurring roughly around Cape Canaveral, FL, is similar to that of other estuarine, marine, and freshwater species with geographic distributions that encompass both regions (Avise 1992; Blum et al. 2007; Drumm and Kreiser 2012). However, unlike in J. roemerianus adjacent genetic clusters in other species are more genetically similar. In his paper examining 19 species across the southeastern United States, Avise (1992) attributed geographic patterns in genetic structure to three possible factors: historical, physical, and climatic. Historical factors may explain both the geographic location of the genetic breaks in J. roemerianus, as well as the pattern of genetic similarity among the clusters. Prior to the emergence of the Florida peninsula in the Miocene epoch (Avise 1992), populations of J. roemerianus in the western Gulf and populations at the upper extent of the southeastern Atlantic coast could have been connected by gene flow into a single admixed population. Constrained gene flow, as indicated by significant pairwise FST measures among all sites, could have allowed the genetic signature from this historical connection to prevail in neutral markers, despite loss of any current gene flow between the SAB and WGC. Genetic distinctiveness of EGC could then be explained by the population being evolutionarily younger than WGC and SAB, potentially also explaining the particular distinctiveness of the peninsular Florida populations (EC1, EC2, EC3) in the PCA. Physical factors, specifically oceanic currents, may also be driving the observed structure in the present. The locations of genetic breaks between the clusters correspond to points at which oceanic currents leave land. The Gulf Stream, a major current in the southeastern United States, deflects away from the coast around Cape Canaveral, FL (Avise 1992), the same location as the genetic break between SAB and EGC. A weaker prevailing marine current deflects away from the coast near sites CS10 and AP (Avise 1992), which may explain the more clinal break between the WGC and EGC clusters. Admixture between western and eastern marsh fragments may still occur in CS5–CS7, while sites CS10 and AP receive far less input from the western Gulf due to this prevailing current. Oceanic currents may also be responsible for the broad scale gene flow occurring to create the three large genetic clusters. On the other hand, observed genetic structure could be driven by adaptation to climatic factors. WGC and SAB clusters are located in temperate regions while the peninsular Florida cluster is within a tropical region, and the location of the transitional zone between climatic regions, again occurs around Cape Canaveral, FL (Avise 1992). Adaptation to different climates may explain the genetic distinctiveness of EGC and the greater similarity between SAB and WGC.
Although use of neutral markers with high mutation rates limits the inferences about timing of events or adaptation, the observed structure is unlikely random. Geographic locations of genetic breaks reflect historic events, oceanic currents and climatic regions, and parallels genetic structure in other species with similar geographic distributions. In the future, a study using more conservative markers, such as cpDNA or mtDNA, would be needed to examine the genetic structure and estimate timelines of genetic divergence in J. roemerianus. Overall, the observed genetic patterns may reflect historical gene flow between western Gulf populations and Atlantic populations, and the genetic similarity is still present due to more recently limited gene flow among marsh fragments and adaptation to similar climates.
Implications for conservation and restoration
Genetic factors such as genetic diversity, clonal diversity, and population structure have vitally important ramifications for restoration (Hufford and Mazer 2003; Kramer and Havens 2009). Our results provide broader context for creating restored populations that reflect natural levels of diversity. Differences in genetic and clonal diversity across the Gulf and Atlantic coasts suggest practitioners may want to focus on different facets of diversity when restoring J. roemerianus on each coast to maintain natural adaptive potential and ecosystem function. Failure to include the higher levels of diversity naturally found in J. roemerianus into restored populations could result in restoration failure in the short term or future die-out due to an inability to adapt to changing environmental and climatic conditions (Kramer and Havens 2009). Genetic diversity will be increasingly important to avoid extinction, especially in coastal plant species that have restricted inland migration due to sea walls and coastal development (Kirwan and Megonigal 2013). Additionally, adequate clonal diversity is important for preventing founder effects such as inbreeding depression, low fitness, and low establishment rates (Mijnsbrugge et al. 2010; Hufford and Mazer 2003), and for preserving ecosystem benefits and processes in restored populations (Reynolds et al. 2012).
Regional differences across coasts in levels of clonal and genetic diversity are potentially indicative of life history adaptation and highlight the need to regionally source transplant stock. Patterns of population structure along each coast may provide further guidance at a finer scale. Although neutral genetic variation does not necessarily correspond to adaptive variation, structure and differentiation detected by molecular markers could still denote locally adapted populations (Hufford and Mazer 2003). Therefore, selecting source populations from within the same genetic cluster may prevent declines in native and restored population fitness associated with outbreeding depression, intraspecific hybridization, and genetic swamping (Hufford and Mazer 2003; Lesica and Allendorf 1999; Kramer and Havens 2009). Genetic clusters offer better guidance for source population selection to restoration practitioners than ecoregions or geographic distance between sites, which are often used to guide selection when genetic data is absent. Restoration success tends to be inversely related to the genetic and environmental distance between source and restored populations, which may not always be well correlated to geographic distance (Montalvo and Ellstrand 2000) as is the case with J. roemerianus. For example, the results of this study suggest that restoration efforts north of Cape Canaveral, FL should source from a northern population to ensure genetically similar stock even if a population to the south is geographically closer. Incorporating the genetic data from this study into coastal restoration practices will help support success in the short term and persistence of restored sites into the future.
Conclusions
Juncus roemerianus is a foundational, regionally dominant species in a declining ecosystem that is in need of further genetic data to adequately inform restoration. A recent study found higher rates of genetic and clonal diversity than expected, suggesting a greater balance between sexual reproduction and clonal propagation than predicted by life history literature (Tumas et al. 2018). Patterns of diversity elucidated through expanding the sample collection to cover a majority of the species range suggests that this balance shifts across ecologically and environmentally divergent coastal regions, potentially indicating that life history strategy varies by environment in J. roemerianus. Population structure across the broader study range demonstrates that geographic proximity may not be indicative of genetic similarity, providing valuable information for transplant selection in restoration efforts. Despite the importance of J. roemerianus to salt marsh function and restoration, there have been limited genetic studies conducted on the species to date, emphasizing the need for a paradigm shift in conservation studies to include widespread, common species alongside the more traditionally studied critically endangered and rare species. Common species such as J. roemerianus and other coastal plant species are increasingly important for ecosystem restoration and must to be investigated now to ensure such species remain common and that the ecosystems they support persist into the future.
Data availability
Sample coordinates and microsatellite genotypes from the current study are available from the corresponding author on reasonable request. Microsatellite markers are archived in NCBI GenBank, accession nos: KX398592–KX398610.
References
Angers B, Magnan P, Plante M, Bernatchez L (2002) Canonical correspondence analysis for estimating spatial and environmental effects on microsatellite gene diversity in brook charr (Salvelinus fontinalis). Mol Ecol 8:1043–1053. https://doi.org/10.1046/j.1365-294x.1999.00669.x
Arnaud-Haond S, Duarte CM, Alberto F, Serrão EA (2007) Standardizing methods to address clonality in population studies. Mol Ecol 16:5115–5139. https://doi.org/10.1111/j.1365-294X.2007.03535.x
Ashley MV (2010) Plant parentage, pollination, and dispersal: how DNA microsatellites have altered the landscape. CRC Crit Rev Plant Sci 29:148–161. https://doi.org/10.1080/07352689.2010.481167
Avise JC (1992) Molecular population structure and the biogeographic history of a regional fauna: a case history with lessons for conservation biology. Oikos 63:62–76
Blum MJ, Jun Bando K, Katz M, Strong DR (2007) Geographic structure, genetic diversity and source tracking of Spartina alterniflora. J Biogeogr 34:2055–2069. https://doi.org/10.1111/j.1365-2699.2007.01764.x
Broome SW, Seneca ED, Woodhouse WW (1988) Tidal salt marsh restoration. Aquat Bot 32:1–22. https://doi.org/10.1016/0304-3770(88)90085-X
Chessel D, Dufour AB, Thioulouse J (2004) The ade4 package-I: one-table methods. R News 4:5–10
Cleary KA, Waits LP, Finegan B (2017) Comparative landscape genetics of two frugivorous bats in a biological corridor undergoing agricultural intensification. Mol Ecol 26:4603–4617. https://doi.org/10.1111/mec.14230
Core Team R (2016) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Cronk JK, Fennessy MS (2001) Wetland plants: biology and ecology. Lewis Publishers, Washington, D.C.
Crutsinger GM, Collins MD, Fordyce JA et al (2006) Plant genotypic diversity predicts community structure and governs an ecosystem process. Cell 647:966–968. https://doi.org/10.1126/science.1128326
Dardeau MR, Modlin RF, Shroeder WW, Stout JP (1992) Estuaries. In: Hackney CT, Adams SM, Martin WH (eds) Biodiversity of the Southeastern United States: aquatic communities. John Wiley & Sons Inc, New York, pp 614–744
Dray S, Dufour A-B (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22:1–20. https://doi.org/10.18637/jss.v022.i04
Dray S, Dufour AB, Chessel D (2007) The ade4 package—I: two-table and K-table methods. R News 7:47–52. https://doi.org/10.1159/000323281
Drumm DT, Kreiser B (2012) Population genetic structure and phylogeography of Mesokalliapseudes macsweenyi (Crustacea: Tanaidacea) in the northwestern Atlantic and Gulf of Mexico. J Exp Mar Biol Ecol 412:58–65. https://doi.org/10.1016/j.jembe.2011.10.023
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Eleuterius LN (1975) The life history of the salt marsh rush, Juncus roemerianus. Bull Torrey Bot Club 102:135–140. https://doi.org/10.2307/2484735
Eleuterius LN (1976) Vegetative morphology and anatomy of the salt marsh rush, Juncus roemerianus. Gulf Res Rep 5:1–10. https://doi.org/10.18785/grr.0502.01
Ellstrand NC, Roose ML (1987) Patterns of genotypic diversity in clonal plant species. Am J Bot 74:123–131. https://doi.org/10.1007/sl2229-008-9001-0
Eriksson O (1989) Seedling dynamics and life histories in clonal plants. Oikos 55:231–238. https://doi.org/10.2307/3565427
Eriksson O (1993) Dynamics of genets in clonal plants. Trends Ecol Evol 8:313–316. https://doi.org/10.1038/hr.2014.17
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508. https://doi.org/10.1046/j.1523-1739.1996.10061500.x
Gabrielsen TM, Brochmann C (1998) Sex after all: high levels of diversity detected in the Arctic clonal plant Saxifraga cernua using RAPD markers. Mol Ecol 7:1701–1708. https://doi.org/10.1046/j.1365-294x.1998.00503.x
Gedan KB, Silliman BR, Bertness MD (2009) Centuries of human-driven change in salt marsh ecosystems. Annu Rev Mar Sci 1:117–141. https://doi.org/10.1146/annurev.marine.010908.163930
Godfrey RK, Wooten JW (1979) Aquatic and wetland plants of the southeastern United States: monocotyledons. University of Georgia Press, Athens
Greenberg R, Maldonado JE (2006) Diversity and endemism in tidal-marsh vertebrates. In: Greenberg R, Maldonado JE, Droege S, McDonald MV (eds) Terrestrial vertebrates of tidal marshes: evolution, ecology, and conservation. Cooper Ornithological Society, Camarillo, pp 32–53
Greenberg R, Maldonado JE, Droege S, McDonald MV (2006) Tidal marshes: a global perspective on the evolution and conservation of their terrestrial vertebrates. Bioscience 56:675–685. https://doi.org/10.1641/0006-3568(2006)56%5b675:TMAGPO%5d2.0.CO;2
Grünwald NJ, Goodwin SB, Milgroom MG, Fry WE (2003) Analysis of genotypic diversity data for populations of microorganisms. Phytopathology 93:738–746. https://doi.org/10.1094/PHYTO.2003.93.6.738
Hämmerli A, Reusch TBH (2003) Inbreeding depression influences genet size distribution in a marine angiosperm. Mol Ecol 12:619–629. https://doi.org/10.1046/j.1365-294X.2003.01766.x
Hijmans RJ (2019) raster: geographic data analysis and modeling. R Package Version 2.8-19. https://CRAN.R-project.org/package=raster
Hufford KM, Mazer SJ (2003) Plant ecotypes: genetic differentiation in the age of ecological restoration. Trends Ecol Evol 18:147–155. https://doi.org/10.1016/S0169-5347(03)00002-8
Hughes AR, Stachowicz JJ (2009) Ecological impacts of genotypic diversity in the clonal seagrass Zostera marina. Ecology 90:1412–1419. https://doi.org/10.1890/07-2030.1
Hughes AR, Inouye BD, Johnson MTJ et al (2008) Ecological consequences of genetic diversity. Ecol Lett 11:609–623. https://doi.org/10.1111/j.1461-0248.2008.01179.x
Huiskes AHL, Koutstall BP, Herman PMJ et al (1995) Seed dispersal of halophytes in tidal salt marshes. J Ecol 83:559–567. https://doi.org/10.2307/2261624
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106. https://doi.org/10.1111/j.1365-294X.2007.03089.x
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr : an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kamvar ZN, Brooks JC, Grünwald NJ (2015) Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front Genet 6:1–10. https://doi.org/10.3389/fgene.2015.00208
Keenan K, Mcginnity P, Cross TF et al (2013) DiveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
Kennish MJ (2001) Coastal salt marsh systems in the U.S.: a review of anthropogenic impacts. J Coast Res 17:731–748. https://doi.org/10.2307/4300224
Kirwan ML, Megonigal JP (2013) Tidal wetland stability in the face of human impacts and sea-level rise. Nature 504:53–60. https://doi.org/10.1038/nature12856
Kramer AT, Havens K (2009) Plant conservation genetics in a changing world. Trends Plant Sci 14:599–607. https://doi.org/10.1016/j.tplants.2009.08.005
Lesica P, Allendorf FW (1999) Ecological genetics and the restoration of plant communities: mix or match? Restor Ecol 7:42–50. https://doi.org/10.1046/j.1526-100X.1999.07105.x
Lloyd MW, Burnett RK, Engelhardt KAM, Neel MC (2011) The structure of population genetic diversity in Vallisneria americana in the Chesapeake Bay: implications for restoration. Conserv Genet 12:1269–1285. https://doi.org/10.1007/s10592-011-0228-7
McKay JK, Christian CE, Harrison S, Rice KJ (2005) “How local is local?”A review of practical and conceptual issues in the genetics of restoration. Restor Ecol 13:432–440. https://doi.org/10.1111/j.1526-100X.2005.00058.x
Mijnsbrugge KV, Bischoff A, Smith B (2010) A question of origin: where and how to collect seed for ecological restoration. Basic Appl Ecol 11:300–311. https://doi.org/10.1016/j.baae.2009.09.002
Montalvo AM, Ellstrand NC (2000) Transplantation of the subshrub Lotus scoparius: testing the home-site advantage hypothesis. Conserv Biol 14:1034–1045. https://doi.org/10.1046/j.1523-1739.2000.99250.x
Narum SR (2006) Beyond Bonferroni: less conservative analyses for conservation genetics. Conserv Genet 7:783–787. https://doi.org/10.1007/s10592-005-9056-y
Novy A, Smouse PE, Hartman JM et al (2010) Genetic variation of Spartina alterniflora in the New York metropolitan area and its relevance for marsh restoration. Wetlands 30:603–608. https://doi.org/10.1007/s13157-010-0046-6
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2019). vegan: Community Ecology Package. R Package Version 2.5-4. https://CRAN.R-project.org/package=vegan
Paine RT, Vadas RL (1969) The effects of grazing by sea urchins, Strongylocentrotus spp., on benthic algal populations. Limnol Oceanog 15:710–719
Pennings SC, Bertness MD (2001) Salt marsh communities. In: Bertness MD, Gaines SD, Hay ME (eds) Marine COMMUNITY ECOLOGY. Sinauer Associates, Sunderland, pp 289–316
Pennings SC, Callaway RM (2000) The advantages of clonal integration under different ecological conditions: a community-wide test. Ecology 81:709–716
Pleuss AR, Stocklin J (2004) Population genetic diversity of the clonal plant Geum reptans (Rosaceae) in the Swiss Alps. Am J Bot 91:2013–2021. https://doi.org/10.3732/ajb.91.12.2013
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Raymond M, Rousset F (1995) Genepop (Version-1.2). Population-genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reusch TBH (2006) Does disturbance enhance genotypic diversity in clonal organisms? A field test in the marine angiosperm Zostera marina. Mol Ecol 15:277–286. https://doi.org/10.1111/j.1365-294X.2005.02779.x
Reusch TBH, Hughes AR (2006) The emerging role of genetic diversity for ecosystem functioning: estuarine macrophytes as models. Estuaries Coasts 29:159–164. https://doi.org/10.1007/BF02784707
Reusch TBH, Ehlers A, Hammerli A, Worm B (2005) Ecosystem recovery after climatic extremes enhanced by genotypic diversity. Proc Natl Acad Sci USA 102:2826–2831. https://doi.org/10.1073/pnas.0500008102
Reynolds LK, McGlathery KJ, Waycott M (2012) Genetic diversity enhances restoration success by augmenting ecosystem services. PLoS ONE 7:1–7. https://doi.org/10.1371/journal.pone.0038397
Richards CL, Hamrick JL, Donovan LA, Mauricio R (2004) Unexpectedly high clonal diversity of two salt marsh perennials across a severe environmental gradient. Ecol Lett 7:1155–1162. https://doi.org/10.1111/j.1461-0248.2004.00674.x
Silvertown J (2008) The evolutionary maintenance of sexual reproduction: evidence from the ecological distribution of asexual reproduction in clonal plants. Int J Plant Sci 169:157–168. https://doi.org/10.1086/523357
Soons MB, Van Der Vlugt C, Van Lith B et al (2008) Small seed size increases the potential for dispersal of wetland plants by ducks. J Ecol 96:619–627. https://doi.org/10.1111/j.1365-2745.2008.01372.x
Stout JP (1984) The ecology of irregularly flooded salt marshes of the northeastern Gulf of Mexico: a community profile. US Fish Wildl Serv Biol Rep 85(71):98. https://doi.org/10.1017/cbo9781107415324.004
Travis SE, Proffitt CE, Ritland K (2004) Population structure and inbreeding vary with successional stage in created Spartina alterniflora marshes. Ecol Appl 14:1189–1202
Tumas HR, Shamblin BM, Woodrey MS, Nairn CJ (2017) Microsatellite markers for population studies of the salt marsh species Juncus roemerianus (Juncaceae). Appl Plant Sci 5:1600141. https://doi.org/10.3732/apps.1600141
Tumas HR, Shamblin BM, Woodrey MS, Nibbelink NP, Chandler R, Nairn CJ (2018) Landscape genetics of the foundational salt marsh plant species black needlerush (Juncus roemerianus Scheele) across the northeastern Gulf of Mexico. Landsc Ecol 33:1585–1601. https://doi.org/10.1007/s10980-018-0687-z
USDA NRCS (2018) The PLANTS database. National Plant Data Team, Greensboro. http://plants.usda.gov
Valiela I, Kinney E, Culbertson J et al (2009) Global losses of mangroves an salt marshes. In: Duarte CM (ed) Global loss of coastal habitats rates, causes and consequences. Fundación BBVA, Madrid, pp 109–142
Widén B, Cronberg N, Widén M (1994) Genotypic diversity, molecular markers and spatial distribution of genets in clonal plants, a literature survey. Folia Geobot Phytotaxon 29:245–263. https://doi.org/10.1007/BF02803799
Williams SL (2001) Reduced genetic diversity in eelgrass transplantations affects both population growth and individual fitness. Ecol Appl 11:1472–1488. https://doi.org/10.1890/1051-0761(2001)011%5b1472:RGDIET%5d2.0.CO;2
Yannic G, Baumel A, Ainouche M (2004) Uniformity of the nuclear and chloroplast genomes of Spartina maritima (Poaceae), a salt-marsh species in decline along the Western European Coast. Heredity 93:182–188. https://doi.org/10.1038/sj.hdy.6800491
Zedler JB, Kercher S (2005) WETLAND RESOURCES: status, trends, ecosystem services, and restorability. Annu Rev Environ Resour 30:39–74. https://doi.org/10.1146/annurev.energy.30.050504.144248
Acknowledgements
The authors thank E. T. Jones, J. Feura, and J. S. Andrews for assistance with field collection. We gratefully acknowledge resources provided by the Warnell School of Forestry and Natural Resources, the University of Georgia Genomics Facility, the Grand Bay National Estuarine Research Reserve (NERR), and Apalachicola NERR.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest:
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tumas, H.R., Shamblin, B.M., Woodrey, M.S. et al. Broad-scale patterns of genetic diversity and structure in a foundational salt marsh species black needlerush (Juncus roemerianus). Conserv Genet 20, 903–915 (2019). https://doi.org/10.1007/s10592-019-01183-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-019-01183-3