
Abstract
As a member of adherens junction, p120-catenin (p120ctn) plays a major role in cell adhesions through stabilization of E-cadherin. p120ctn is transcriptionally down-regulated in non-small cell lung cancer (NSCLC), although the molecular mechanisms underlying p120ctn repression are incompletely defined. Here we further investigated transcriptional regulation of p120ctn in NSCLC. We prepared a promoter reporter plasmid construct that contained p120ctn promoter region from position −1082 to +320 relative to transcription start site. Through serial deletion mutation analysis of the p120ctn promoter, we pinpointed cis-acting elements involved in regulation of p120ctn. We identified transcription factor SP1 as a transcriptional repressor of p120ctn that directly binds to segment (−9 to +36) of the p120ctn promoter. SP1 can receive multiple signals from several intracellular signaling pathways. Through examination of SP1 binding partners, we identified proto-oncogene c-Crk to be involved in transcriptional down-regulation of p120ctn. RNAi mediated silencing of CRK in A549, H157 and H358 cells increased p120ctn protein levels. On the other hand, over-expression of CRK-I and CRK-II in NSCLC cells down-regulated p120ctn, an effect that was abrogated by simultaneous silencing of SP1. In summary, our data provide evidence for the role of c-Crk proto-oncogene in transcriptional repression of p120ctn that further clarifies the mechanism by which this biochemical signal promotes metastasis in NSCLC.
Similar content being viewed by others


Introduction
As a member of adherens junction, the p120-catenin protein (p120ctn) plays an important role in cell–cell adhesions. Loss of p120ctn expression results in destabilization of the E-cadherin complex thereby promoting tumor invasion and metastasis [3, 5, 6, 9, 11, 23, 24, 29–31]. Down-regulation of p120ctn has been shown to disrupt cell–cell adhesions in several studies [6, 9, 21, 29, 30]. p120ctn knockdown by RNA interference results in dose-dependent elimination of epithelial, placental, neuronal and vascular endothelial cadherins and complete loss of cell–cell adhesion [6]. Moreover, decreased expression of p120ctn is observed in multiple tumors types and is associated with increased metastatic potential of these tumors including 60–80% of all subtypes of NSCLC [5, 12–15, 24, 27, 29, 31].
Decreased expression of p120ctn in NSCLC is most commonly due to transcriptional down-regulation of the p120ctn gene [12, 15, 19]. We recently cloned the p120ctn promoter into a firefly luciferase reporter vector and observed that promoter activity of p120ctn is significantly reduced in NSCLC cell lines compared to immortalized normal human respiratory epithelial cells [19]. Through serial deletion analysis of the p120ctn promoter, we identified two separate regions of this promoter both of which hold significant transcriptional regulatory functions [i.e., segments (+267 to +282) and (−9 to +36) relative to transcription initiation site]. We recently published the results of p120ctn promoter analysis between positions +267 and +282 and reported the role of FOXC2 (mesenchymal forkhead related protein) in transcriptional down-regulation of the p120ctn [19]. Here, we report our analysis results of segment (−9 to +36) of the p120ctn promoter. We identified transcription factor SP1 as another regulator of the p120ctn transcription which directly binds to segment (−9 to +36) of this promoter.
Specificity protein 1 (SP1) belongs to a universally expressed zinc-finger DNA binding proteins that are involved in transcriptional regulation of many genes [26]. Multiple proteins including other transcription factors can interact with SP1 and thereby transduce their respective signal for gene regulation through SP1. We examined several binding partners of SP1 and recognized the role of the c-Crk proto-oncogene in transcriptional down-regulation of the p120ctn.
Materials and methods
Cell cultures
A549, H157 and H358 cells were routinely cultured in RPMI supplemented with antibiotics and 10% heat-inactivated FBS (Omega Scientific, Tarzana, CA). Immortalized normal human epithelial cells (BEAS-2B) were cultured in BEBM medium supplemented with all the additives (Lonza/Clonetics Corporation, Switzerland; Catalog No. CC-3170).
Measurement of p120ctn promoter activity by dual luciferase assay
p120ctn promoter constructs were transfected into NSCLC cell lines as well as immortalized normal human respiratory epithelial cells (BEAS-2B) by Lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection cells were washed with PBS and lysed using a Branson Sonifier in 1× passive lysis buffer (Promega) at room temperature (RT). Reporter gene expression was assessed by using the Dual-Luciferase Reporter Assay System kit (Promega) according to manufacturer’s instructions in a TD-20/20 Luminometer (Turner Biosystems, Sunnyvale, CA). We normalized for transient transfection efficiency (i.e. firefly luciferase activity) by co-transfection of a Renilla luciferase expressing control vector (pRL-SV40). All experiments were performed in triplicate and were reported as means ± standard deviation (s.d.), and each experiment was performed at least twice.
siRNA mediated gene silencing
Initially, A549 cells were transfected with two independent sets of siRNA for p120ctn, AP2, SP1, and CRK. In the case of NF-1 we chose one set of siRNA since there was no significant correlation between NF-1 silencing and the p120ctn promoter activity. Transfections were performed by using Lipofectamine 2000 (Invitrogen).
The siRNA duplex sequences that we used to silencing the above mentioned genes are as follows:
p120ctn #1:
5′-GCUAUGAUGACCUGGAUUAdtdt-3′
5′-UAAUCCAGGUCAUCAUAGCdtdt-3′
p120ctn #2:
5′-CUAUGAUGACCUGGAUUAUdtdt-3′
5′-AUAAUCCAGGUCAUCAUAGdtdt-3′
AP2 #1:
5′-GCCAAAGCAGUAGCUGAAUdtdt-3′
5′-AUUCAGCUACUGCUUUGGCdtdt-3′
AP2 #2:
5′-GAUCAAACUGUAAUUAAGAdtdt-3′
5′-UCUUAAUUACAGUUUGAUCdtdt-3′
SP1 #1:
5′-GCAACAUGGGAAUUAUGAAdtdt-3′
5′-UUCAUAAUUCCCAUGUUGCdtdt-3′
SP1 #2:
5′-CAUACCAGGUGCAAACCAAdtdt-3′
5′-UUGGUUUGCACCUGGUAUGdtdt-3′
NF1 #1:
5′-CCAAAGACGUGGUUGAUGAdtdt-3′
5′-UCAUCAACCACGUCUUUGGdtdt-3′
CRK #1:
5′-CUGCUUACCCUGAUUUAUUdtdt-3′
5′-AAUAAAUCAGGGUAAGCAUdtdt-3′
CRK #2:
5′-CCUGAUUUAUUCAGUGGUUdtdt-3′
5′-AACCACUGAAUAAAUCAGGdtdt-3′
As a control, scrambled non-silencing siRNA was used (Sigma-Aldrich Cat number SIC002-10NMOL). Concentrations of siRNA were kept at 100 nM among groups.
Measurement of mRNA level by RT-PCR
We extracted total RNA from each NSCLC cell line by using Trizol method and prepared cDNA using SuperScript-II RT Kit (Invitrogen) and Oligo (dT)s according to the manufacturer’s instructions. The following primer sets were used to amplify a short segment of cDNA for each gene.
AP2-forward:
5′-TATCTACCCCCAGTCGCAAG-3′
AP2-reverse:
5′-TACCCGGGTCTTCTACATGC-3′
NF-1-forward:
5′-GCCATTCTCAAGAGGCAGTC-3′
NF-1-reverse:
5′-CATTCGTATTGCTGGGTGTG-3′
SP1-forward:
5′-GGAGAGCAAAACCAGCAGAC-3′
SP1-reverse:
5′-CAATGGGTGTGAGAGTGGTG-3′
MYC-forward:
5′-CTCCTGGCAAAAGGTCAGAG-3′
MYC-reverse:
5′-GGCCTTTTCATTGTTTTCCA-3′
E2F-1-forward:
5′-GCCAAGAAGTCCAAGAACCA-3′
E2F-1-reverse:
5′-CTCAGGGCACAGGAAAACAT-3′
SRC-forward:
5′-CTGTTCGGAGGCTTCAACTC-3′
SRC-reverse:
5′-TGAGAGGCAGTAGGCACCTT-3′
CRK forward:
5′-GCAAGAGAGGGATGATTCCA-3′
CRK reverse:
5′-ATGGGAAGTGACCTCGTTTG-3′
Actin-forward:
5′-TGACGGGGTCACCCACACTGTGCCC
ATCTA-3′
Actin-reverse:
5′-CTAGAAGCATTTGCGGTGGACGATG
GA-3′
All PCR reactions were performed by using Taq DNA polymerase (Roche Applied Science) in an Eppendorf Mastercycler thermo cycler (Eppendorf).
Measurement of mRNA level by qRT-PCR
We used TaqMan probes and SYBR Green methods for measurement of mRNA level following gene silencing. In case of p120ctn, mRNA level in NSCLC cell lines were measured by TaqMan probes; assay IDs: Hs00931670_m1 and Hs00183425_m1 (Applied Biosystems, Foster City, CA). For CRK, AP2, NF-1 and SP1 we used SYBR Green method with the above mentioned primers.
In each cell type, total RNA was extracted by using Trizol method and cDNA was prepared by an RT-PCR reaction using SuperScript-II RT Kit (Invitrogen) and Oligo (dT)s according to the manufacturer’s instructions. For each cDNA sample an internal control (beta actin) was also measured by TaqMan probe. In order to compare the expression level of mRNA between samples, we used the Comparative Ct Method (ΔΔCt). Relative expression of mRNA compared to beta actin in each sample was calculated (ΔCt) and relative expression of mRNA among samples was determined by calculating the difference in (ΔCt) between samples (ΔΔCt). All relative quantitative PCRs were performed, recorded, and analyzed by using the ABI 7300Prism Sequence Detection System (Applied Biosystems, Foster City, CA). All samples were carried out in triplicate (10 ng of total RNA per well) and repeated at least twice. Controls without template or reverse transcriptase were run in each experiment.
Western blots
NSCLC cell lines were seeded in 10 cm Petri dishes at 5 × 105 cells per dish, which resulted in 30–40% confluency 24 h after plating. Cells were harvested at 24 h by adding trypsin, pelleted and lysed in 100 μl of lysis buffer (NaCl 15 mM; EDTA 0.5 mM; Tris 10 mM) using a Branson Sonifier. Cell debris was collected by centrifugation at 4°C, and protein concentration was measured by the BCA method. Protein was resolved by SDS-PAGE and was transferred to a nitrocellulose membrane. The membrane was blocked with TBS with 5% nonfat powdered milk.
Membranes were immunoblotted with the following primary antibodies: p120ctn (BD biosciences Cat. number 610133); SP1 (Abcam Cat. number 58199); E-cadherin (BD biosciences Cat. number 61081); CRK II (Santa Cruz Biotechnology Cat. number sc-289); CRK I/II (Santa Cruz Biotechnology Cat. number sc-17989). Horse radish peroxidase conjugated secondary antibodies were used for detection of bands by chemiluminescence (ECL western blotting detection reagents, Amersham Biosciences, Piscataway, NJ, USA).
Chromatin immunoprecipitation assay (ChIP)
We followed the method described by Boyd and Farnham [2] and Y. Shang et al. [25] with some modifications. 1. A549 and H157 cells (2 × 107 in a 150 mm dish) were grown at confluence. 2. Cells were cross-linked by adding formaldehyde to a final concentration of 1% (0.68 ml of 37%/25 ml media) directly into the media and mixed on a rocker for 10 min at room temperature. 3. The cross-linking was stopped by adding glycine to a final concentration of 125 mM (3.75 ml of 1 M/25 ml media) for 5 min at room temperature. 4. The cell monolayers were washed three times with ice-cold 1× PBS. Cells were then scraped into 1× PBS (5 ml) plus protease inhibitors and collected by centrifugation (700×g for 4 min). 5. Cell pellets were re-suspended in cell lysis buffer [5 mM Pipes (KOH), pH 8.0/85 mM KCl/0.5% NP-40] containing the following protease inhibitors 1 μg/ml leupeptin, 1 μg/ml aprotinin and 1 mM PMSF and incubated for 10 min on ice. The efficiency of cell lysis was checked with trypan blue and if needed cells were homogenized on ice with a type B homogenizer. 6. Nuclei were pelleted by centrifugation (5000 rpm for 5 min) and re-suspend in nuclear lysis buffer [50 mM Tris, pH 8.1/10 mM EDTA/1% SDS containing the same protease inhibitors as in cell lysis buffer] followed by incubation on ice for 10 min. 7. Chromatin was shredded by using a sonifier to an average length of about 600 bp while keeping samples on ice. For both cell lines, we used a Branson Sonifier 250 with power setting 5 in 20-s. bursts followed by 1 min of cooling on ice for a total sonication time of 3 min per sample. 8. Debris was cleared by centrifugation at maximum speed for 10 min at 4°C. 9. The supernatant was transferred to a new tube and dilute 5-fold in ChIP dilution buffer [0.01% SDS/1.1% Triton X-100/1.2 mM EDTA, 16.7 mM Tris, pH 8.1/167 mM NaCl plus protease inhibitors]. 10. To reduce nonspecific background, we pre-cleared the samples with 80 μl of salmon sperm DNA/protein A/G agarose slurry for 30 min at 4°C with agitation. 11. We collected the beads by a brief centrifugation and separated the supernatant fraction. 12. 20% of the total supernatant was saved as total input control and processed with the eluted Immunoprecipitates. 13. The rest of the supernatant was divided into two fractions: one for an IP with IgG control and the second was incubated with 5 μg of SP1 antibody (Abcam Cat. number ab13405) overnight at 4°C with rotation. 14. We collected immune complexes with 60 μl of the salmon sperm DNA/protein A/G agarose slurry for 1 h at 4°C with rotation. 15. Beads were then washed consecutively for 3–5 min on a rotating platform with 1 ml of each solution:
Low salt wash buffer [0.1% SDS/1% Triton X-100/2 mM EDTA, 20 mM Tris, pH 8.1/150 mM NaCl] b. high salt wash buffer [0.1% SDS/1% Triton X-100, 2 mM EDTA, 20 mM Tris, pH 8.1, 500 mM NaCl] c. LiCl wash buffer [0.25 M LiCl/1% NP40/1% deoxycholate, 1 mM EDTA/10 mM Tris, pH 8.0] d. twice in 1× TE buffer. 16. Complexes were eluded by adding 250 μl of elution buffer [1% SDS/0.1 M NaHCO3] to pelleted beads by mixing and shaking by vortexer for at least 15 min. Beads and eluted proteins were separated by centrifugation at 14,000 rpm for 3 min. This step was repeated and both elutants were combined in the same tube. 17. Formaldehyde cross links were reversed by adding 1 μl 10 mg/ml RNase and 5 M NaCl to a final concentration of 0.3 M to the elutants and incubation in a 65°C for 4–5 h. 18. DNA was precipitated by adding 2 1/2 volumes of 100% ethanol overnight at −20°C. 19. DNA and debris were collected by centrifugation at high speed and re-suspend in 100 μl of water. We added 2 μl of 0.5 M EDTA, 4 μl 1 M Tris, pH 6.5 and 1 μl of 20 mg/ml Proteinase K and incubated samples for 1–2 h at 45°C and purified DNA by using QiaQuick spin columns. 20. 2 μl of this DNA samples was used in qRT-PCR reaction by SYBR Green with the following primers.
SP1 ChIP-forward:
5′-GGTTCCTATTGGAAGCTCAC-3′
SP1 ChIP-reverse:
5′-CTCTCTCTCTCCCTCT-3′
Immunoprecipitation
We grew A549 and H157 cells in 100 cm2 dishes to 90% confluency. Cells were washed with 2 ml PBS and scraped off in 1 ml PBS. Cells were transferred to Eppendorf tubes and spun at 1000 RPM at 4°C for 10 min. Then we prepared a cell lysate by re-suspending the cells in ice-cold gentle lysis buffer [10 mM Tris–HCl pH 7.5; 10 mM NaCl; 2 mM EDTA; 0.1% Triton-X100; 1 mM PMSF; 2 μg/ml aprotinin; 2 μg/ml leupeptin] (approximately 700 μl per 2 × 106 cells). Cells were incubated on ice for 5 min before adding NaCl to 150 mM followed by incubating on ice for 10 min. Next, cells were spun again at 14,000 rpm in 4°C for 15 min. We split the supernatant in two fractions and incubated them with either 4 μg of anti CRK antibody (Santa Cruz Biotechnology Cat. number sc-289); SP1 (Abcam Cat. number 13405) or 4 μg of control IgG for 4 h. Subsequently, we added 25 μl of protein G plus/protein A agarose suspension (Calbiochem Cat. number IP05) and incubated overnight at 4°C with agitation. We washed the beads 8 times with 1 ml of ice cold NET [50 mM Tris–HCl pH 7.5; 150 mM NaCl; 0.05% Triton-X100] for 1 min each time spinning at 1,000 rpm at 4°C. Eventually, we eluted the immunoprecipitate by adding SDS directly to beads and proceeded with western blotting with mouse anti SP1 antibody (Abcam Cat. number ab58199).
CRK over-expression
We obtained a CRK-I expression plasmid and its corresponding empty control vector from (OriGene Technologies Cat number sc109105 and PCMV6XL5) as well as CRK-II expression plasmid (OriGene Technologies Cat number RC201701 and PCMV6-Entry). For CRK-I, H358 cells were transiently transfected with the above mentioned plasmids by using Lipofectamine 2000 (Invitrogen). Six hours after transfection, cells were washed and transfected again with appropriate siRNA (SP1 or non-silencing siRNA). In case of CRK-II, A549 cells were transfected with the appropriate plasmids and 48 h post transfection, cells were selected by G418 at 1000 μg/ml for 2 weeks. We used 1.0 μg of plasmid DNA for every well in a 6 well plate format. Following selection of stably transfected cells, these cells were transfected with appropriate siRNA (SP1 or non-silencing siRNA). Cells were kept in 37°C incubator and were analyzed by western blotting at 48 h following siRNA transfection.
Results
Promoter activity of p120ctn is significantly reduced in NSCLC cell lines compared to immortalized normal human respiratory epithelial cell line (BEAS-2B)
We recently reported that altered expression of p120ctn was attributable to transcriptional down-regulation of the p120ctn promoter [19]. We compared p120ctn promoter activity in NSCLC cell lines to that of an immortalized normal respiratory epithelial cell line, BEAS-2B. BEAS-2B cells represent human bronchial epithelial cells infected by the SV40 virus, which results in a non-tumorigenic immortalized cell line. We amplified promoter region of the p120ctn spanning from position −1082 to +320 relative to transcriptional start site and cloned the resulted amplicon into a firefly luciferase reporter vector (pGL4.16 Luciferase reporter vector, Promega) to generate p120ctn short-luc. Our p120ctn promoter construct was transiently transfected into several NSCLC cell lines as well as BEAS-2B cells and reporter gene activity was measured at 24 h. The p120ctn promoter activity in all tested NSCLC cells was significantly lower than BEAS-2B cells (Fig. 1a). This finding suggests that differential transcriptional regulation of p120ctn in malignant versus non-malignant lung epithelial cells contributes to lower expression level of p120ctn in malignant lung epithelium.

a Promoter analysis of p120ctn in A549 (NSCLC) and BEAS-2B (immortalized normal human epithelium) cell lines. Results are normalized for transient transfection efficiency (i.e. firefly luciferase activity) by co-transfection of a Renilla luciferase expressing control vector (pRL-SV40). Numbers on horizontal axis indicate quantity of DNA transfected. Promoter activity of A549 cells are significantly reduced compared to BEAS-2B cells. b Analysis of p120ctn promoter by creating serial deletion constructs. These deletion constructs of full length p120ctn promoter were prepared by deletions from both 5′ and 3′ ends of the p120ctnshort-luc construct. Both A549 and BEAS-2B cells were transfected with this mutant constructs and 24 h later cells were lysed and subjected to dual Luciferase assay, data only shown for A549 cells. Relative promoter activity is normalized to full length promoter activity in A549 cells. p120ctn promoter activity significantly changes upon deletion segments (−9 to +36) and (+267 to +320). c Chromatin immunoprecipitation (ChIP) in A549 and H157 cells; immunoprecipitation with anti-SP1 antibody or IgG control, PCR with primers encompassing segment (−56 to +111) of the p120ctn promoter. Error bars represent 95% confidence interval.SP1 binds to the p120ctn promoter. d Schematic view of the p120ctn core promoter region and putative cis-acting elements both at the 5′ and 3′ sides of transcription initiation site that were predicted by Alibaba2 analysis. Binding sites for transcription factors NF-1, AP-2 and SP1 are predicted within segment (−9 to +36) of the p120ctn promoter
Two distinct regions of p120ctn promoter harbor significant transcriptional regulatory elements
Considering the finding of reduced p120ctn promoter activity in NSCLC cells, we next sought to identify additional regulatory elements that mediate transcriptional repression of the p120ctn in malignant lung epithelium. In order to identify the important and relevant sequences in the p120ctn promoter region involved in p120ctn transcriptional down-regulation, we performed serial deletions of our p120ctn short-luc promoter construct.Footnote 1 Specifically, we created 3′ deletion mutant constructs spanning from position −221 to positions +267, +239, +69, +19, −9, −20 and −71 relative to transcription initiation site. The schematic view of the deletion mutant constructs and their relative promoter activity compared to full length promoter in A549 cells are shown in (Fig. 1b).
Deletion of the first 53 nucleotides from the 3′ end of our p120ctn short-luc promoter construct, [mutant (−221 to +267)] resulted in a robust increase in promoter activity compared to full length construct. Detailed analysis of this segment of the p120ctn promoter revealed that transcription factor FOXC2 (mesenchymal forkhead related protein) in fact binds to segment (+267 to +282) of the p120ctn promoter and negatively regulates this gene [19].
Further 3′ deletions of p120ctn short-luc construct revealed other key regulatory regions of p120ctn promoter. Noticeably, sequential 3′ deletion of segment (−9 to +36) of the p120ctn short-luc construct resulted in a dramatic decrease in luciferase activity [mutants (−221 to −9); (−221 to +19) and (−221 to +36)] (Fig. 1b). This significant loss of promoter activity, identifies segment (-9 to +36) of the p120ctn as part of the core promoter region of this gene which could contain repressive or activating cis-acting elements. Interestingly, upon deletion of segment (-9 to +36), most of the observed difference in promoter activity between A549 and BEAS-2B cells disappeared [mutant (−221 to −9)] (Fig. 2b). This finding suggests that the residing cis-acting elements within this segment of the p120ctn promoter mediate the observed difference in the p120ctn promoter activity between BEAS-2B and A549 cells.

a RT-PCR showing silencing of AP-2, NF-1 and SP1 in A549 and BEAS-2B cells. b Relative p120ctn promoter activity in A549 and BEAS-2B cells following silencing AP2, NF-1 and SP1. Two separated p120ctn promoter constructs [i.e., constructs (−221 to −9) and (−221 to +36)] were used in this analysis. p120ctn promoter activity significantly increases following silencing SP1 in A549 cells to the level seen in BEAS-2B cells
In order to identify putative cis-acting elements that regulate p120ctn promoter, we used a computer based DNA sequence analysis program known as AliBaba2, a software program for predicting binding sites of transcription factors in an unknown DNA sequence utilizing the TRANSFAC database [8]. Analysis of the p120ctn promoter region in the vicinity of segment (−9 to +36) predicted binding sites for the following transcription factors: NF-1, AP2 and SP1 as well as a NFκB site, immediately 5′ to this segment (Fig. 1d).
Transcription factor SP1 mediates transcriptional regulation of p120ctn
In order to identify the relevant cis-acting elements within the segment (−9 to +36) of p120ctn promoter, we chose to examine the corresponding transcription factor for each predicted cis-acting element. For this purpose, we silenced each putative transcription factor by RNA interference. We obtained separate siRNA sets directed against transcription factors AP2, NF-1and SP1 and tested the silencing efficiency of each siRNA separately (Supplemental Fig. 1). Next we examined the p120ctn promoter activity in A549 and BEAS-2B cells following silencing the above mentioned transcription factors. We used two separate p120ctn promoter constructs [i.e., constructs (−221 to −9) and (−221 to +36)] for this analysis. The difference of promoter activity between these two constructs would be a reflection of segment (−9 to +36) role in transcriptional regulation of the p120ctn. Compared to scrambled control siRNA, SP1 silencing resulted in a robust increase in p120ctn promoter activity in A549 cells, whereas silencing NF-1 had no effect on promoter activity and AP2 silencing resulted in a modest increase in p120ctn promoter activity that was substantially less than that observed for SP1 silencing (Fig. 2).
It is noteworthy that baseline promoter activity and expression level of p120ctn is higher in non-malignant, immortalized BEAS-2B cells compared to NSCLC cell lines including A549 cells [19]. Importantly, the p120ctn promoter activity following SP1 silencing led to a reversal of the p120ctn promoter activity pattern with greater activity in A549 cells compared to BEAS-2B cells. In order to test the generalizability of the SP1 role in the regulation of p120ctn, we examined p120ctn promoter activity following silencing SP1 in two other NSCLC cell lines. We also observe increasing p120ctn promoter activity and p120ctn protein level following silencing transcription factor SP1 in H358 and H157 cells (Fig. 3). The above findings strongly point to the fact that transcription factor SP1 is involved in transcriptional repression of p120ctn.

Relative p120ctn promoter activity and protein level following silencing SP1 in A549, H157 and H358 NSCLC cell lines. Two separate p120ctn promoter constructs [i.e., constructs (−221 to −9) and (−221 to +36)] were used for the promoter analysis. p120ctn promoter activity and protein levels are increased in several NSCLC cells following SP1 silencing
SP1 directly binds to p120ctn promoter
In order to confirm the direct binding of SP1 to the segment (−9 to +36) of the p120ctn promoter, we investigated this DNA–protein interaction by chromatin immunoprecipitation assay (ChIP). As described in the materials and methods section, a 25 μg chromatin sample of A549 and H157 NSCLC cells were prepared and incubated with SP1 antibody or control IgG. Following immunoprecipitation and appropriate washing steps, harvested DNA was used in a real-time PCR reaction to amplify a short 167 bp segment of the p120ctn promoter from position −56 to +111 encompassing the segment (−9 to +36) (Fig. 1c). We observed that a SP1 antibody successfully immunoprecipitated fragments of the p120ctn promoter, which contain segment (−9 to +36) thereby confirming the direct binding of SP1 to the endogenous p120ctn promoter in these two NSCLC cell lines.
CRK expression level in NSCLC cells in inversely correlated with p120ctn expression
Specificity protein 1 (SP1) belongs to a family of universally expressed zinc-finger DNA binding proteins, which are involved in transcriptional regulation of a variety of genes. Multiple signaling pathways transduce their signals through interactions with SP1 including EGFR-through ERK2, cyclin-dependent kinase-2 (CDK2), protein kinase C, HGF- through PI3 kinase and TGF-β -through SMAD2/3 [4]. Moreover, several other proteins including oncoproteins, such as c-Abl, c-Crk, c-Myc, E2F-1 and c-Src, make binary interactions with SP1 [28]. Therefore, we reasoned that studying protein–protein interactions that involve SP1 would provide insight into the abnormal signals that mediate transcriptional down-regulation of p120ctn in NSCLC. For this purpose we examined the expression levels of SP1 and several SP1-interacting oncogenes and correlated them with the expression level of p120ctn in several NSCLC cell lines as well as BEAS-2B cells (Fig. 4). Specifically, we compared mRNA levels of CRK, MYC, E2F-1, SRC and ABL in a panel of NSCLC and BEAS-2B cells by RT-PCR (Fig. 4b). We did not observe a linear correlation between SP1 expression level and p120ctn in our panel of cells. Two cell lines with higher E-cadherin and p120ctn (i.e., BEAS-2B and H358) demonstrate slightly higher SP1 levels (Fig. 4a) but H157 cells with the lowest E-cadherin and p120ctn levels in our panel, demonstrates an SP1 protein level that is identical to A549 and Rh2 cells both of which with higher p120ctn levels. Meanwhile we observed a reverse correlation between the mRNA level of CRK-I/II and p120ctn protein expression in our panel of NSCLC and BEAS-2B cells. This correlation was not seen between p120ctn protein expression and mRNA levels of MYC, SRC, E2F-1 and ABL (data not shown for ABL). In case of SRC, our data shows a reverse correlation between SRC mRNA levels and p120ctn expression in a subset of examined cells. In order to confirm our RT-PCR data, we also measured CRK mRNA levels in our panel of cell lines by real-time qRT-PCR (Fig. 4c). Again, the same reverse correlation between total CRK mRNA level and p120ctn protein expression was observed. We also checked CRK-I/II protein expression in our panel of NSCLC cell in a western blot (Fig. 5). Again, NSCLC cells with lower p120ctn expression, (i.e., Rh2 and H157) demonstrate higher levels of CRK-I/II which is in concordance with our RT-PCR data. The CRK family of adaptor proteins (i.e., CRK-I, CRK-II and their paralogue CRKL) are cellular homologues of v-Crk (CT10 avian sarcoma oncogene) that are involved in transduction of many intracellular signals. These data suggest that SP1’s role in the transcriptional regulation of p120ctn is transmitting the suppressive signal of CRK oncoprotein.

a Western blots showing the expression level of E-cadherin, p120ctn and SP1 in a panel of NSCLC and BEAS-2B cells. b Conventional RT-PCR analysis of mRNA for candidate SP1 binary interaction partners including CRK, MYC, E2F and SRC in the above mentioned panel of cells. c Quantitative RT-PCR measuring CRK mRNA levels in our panel of NSCLC as well as BEAS-2B cells. CRK mRNA levels inversely correlates with p120ctn levels in NSCLC cells

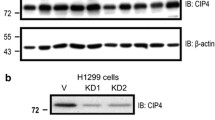
a Western blots showing the expression levels of CRK-I/II in a panel of NSCLC cells. b Conventional RT-PCR measuring mRNA expression level of CRK-I/II in these NSCLC cells. Cell lines with lower p120ctn show higher CRK-I/II mRNA and protein levels
Silencing CRK results in increased p120ctn promoter activity, mRNA and protein levels
Our earlier results are suggesting a role for proto-oncogene c-Crk in transcriptional repression of the p120ctn therefore, we proceeded to examine whether alteration in CRK level would affect the p120ctn promoter activity and protein expression levels. Specifically, we measured p120ctn promoter activity, mRNA and protein levels following siRNA mediated silencing of CRK in three NSCLC cell lines with high, intermediate and low CRK levels.Footnote 2 p120ctn mRNA levels measured by real time qRT-PCR is increased by ~2.5-fold in H157 and A549 cells following siRNA mediated silencing of CRK (Fig. 6). This increase in p120ctn mRNA transcripts was modest in H358 cells that have lower levels of CRK at baseline. We also measured p120ctn promoter activity following siRNA mediated silencing of CRK in H157, A549 and H358 NSCLC cells. Again a pattern of increasing p120ctn promoter activity is observed following silencing CRK which is analogous to the baseline CRK level in these cells (Fig. 6b). In addition to p120ctn promoter activity and mRNA level, we also examined p120ctn protein level following CRK silencing by siRNA in the above mentioned cell lines by western blotting (Fig. 7). Relative to non-silencing siRNA, all examined cell lines show increasing levels of p120ctn following siRNA mediated silencing of CRK, but again the relative increase was muted in H358 cells that manifest lower CRK levels at baseline.

a p120ctn mRNA levels measured by qRT-PCR following silencing CRK in A549, H157 and H358 cells. bp120ctn promoter activity in the above mentioned cells following silencing CRK. c qRT-PCR showing the efficiency of CRK silencing in these cell lines. (2 tailed student’s t-test: * P < 0.0029; ** P < 0.00006; *** P < 0.011; error bars represent ±standard deviation). Silencing CRK, results in increased p120ctn promoter activity and RNA transcripts

Western blots measuring p120ctn protein level in A549, H157 and H358 cells following silencing CRK. In order to assess the specificity of our RNAi, we also silenced p120ctn in these cells. Silencing CRK, lead to an increase in p120ctn protein levels in several NSCLC cell lines
SP1 directly binds to CRK
An interaction between Sp1 and CRK has been proposed through a peptide array screening [28]. A group of 12 Src homology (SH3) domains from eight human proteins including CRK were used to screen for possible interactions of these proteins in a peptide target array composed of 1536 potential ligands. This screening led to the identification of multiple binary interactions between these proteins and their targets including the interaction between SP1 and CRK. Nevertheless, the SP1-CRK interaction has not been directly investigated to the best of our knowledge. Therefore, we sought to examine this protein–protein interaction in a co-immunoprecipitation assay. Indeed, CRK-II co-immunoprecipitated with SP1 following transient transfection of CRK-II-myc in 293-T cells and immunoprecipitation with SP1 antibody (Fig. 8a). In addition, co-immunoprecipitation of endogenous CRK-II and SP1 was observed in Rh2 NSCLC cells (Fig. 8b). In the reverse experiment, we detected SP1 in the immunoprecipitate of A549 and H157 cells isolated with an anti-CRK-II antibody (Fig. 8c). These successful co-immunoprecipitations, confirm the interaction between SP1 and CRK-II which most likely takes place through (SH3) domains of CRK-II.

a Co-immunoprecipitation of CRK-II-myc by SP1 in 293T cells following expression of CRK-II-myc. CRK-II co-immunoprecipitates with SP1. b Co-immunoprecipitation of endogenous CRK-II by SP1 in Rh2 NSCLC cells. Endogenous CRK-II immunoprecipitates with SP1. c Co-immunoprecipitation of SP1 by CRK-II in A549 and H157 cells. Immunoprecipitation was performed with a rabbit anti-CRK-II and western blots by a mouse anti-SP1 and rabbit anti CRK-II. SP1 co-immunoprecipitates with CRK-II in NSCLC cells
CRK effect on p120ctn expression is mediated through SP1
To further establish a causal relationship for the CRK and SP1 interaction in p120ctn transcriptional repression, we examined the effects of concurrent SP1 and CRK manipulation on the p120ctn expression. As shown in Figs. 3, 6 and 7, silencing either SP1 or CRK resulted in increased levels of p120ctn. Here we examined the effects of CRK over-expression with or without SP1 silencing to establish that SP1 is epistatic to CRK in the regulation of p120ctn expression. Stable expression of CRK-II in A549 NSCLC cells, resulted in lower p120ctn protein level and promoter activity (Fig. 9). Silencing SP1 in these cells relieved the CRK-II mediated p120ctn transcriptional repression and restored p120ctn protein levels. Moreover, ectopic expression of CRK-I in H358 NSCLC cells also resulted in a decrease in p120ctn protein levels. We also observed the relief of CRK-I mediated p120ctn suppression upon silencing SP1 in these cells. (Supplemental Fig. 2) Taken together, these results indicate that SP1 functions downstream of CRK vis-à-vis p120ctn transcriptional regulation.

ap120ctn promoter activity following siRNA mediated silencing of Sp1 in A549 cells which were stably transfected with CRK-II or empty vector. b Western blots showing p120ctn protein level in A549 cells with stable expression of CRK-II following siRNA mediated silencing of SP1. CRK-II mediates downregulation of p120ctn which is relieved by simultaneous SP1 silencing
Discussion
The role of p120ctn in the stability of adherens junctions is well established. Down-regulation of this protein in several tumor types including NSCLC [14, 15, 23, 24, 27, 31] results in heightened tumor invasion and metastasis. Thus, the identification of pathways that participate in the down-regulation of p120ctn will elucidate the underlying mechanisms that drive the metastatic process.
Decreased expression of p120ctn in NSCLC seems to be the result of transcriptional down-regulation of this gene [19]. By cloning and analysis of p120ctn promoter, we have identified two regions within this promoter that effectively regulate p120ctn expression. We found that transcription factors FOXC2 (Fork head related protein) and SP1 directly bind to segments (+267 and +282) and (−9 to +36) of the p120ctn promoter respectively and mediate transcriptional repression of this gene. Even though, segment (−9 to +36) of the p120ctn promoter has an overall activating effect on the transcription of this gene, deletion of segment (−9 to +36) abolishes the observed difference in the p120ctn promoter activity between malignant (A549) and non-malignant (BEAS-2B) respiratory cells. Therefore, it seems that both activating and repressive transcription factors bind to segment (−9 to +36) and the sum of their effect on the p120ctn transcription is a net activating result. Among the transcription factors that bind to segment (−9 to +36), SP1 plays a repressive role manifested as an increase in the p120ctn promoter activity upon silencing SP1. Other segments of the p120ctn promoter also seem to have significant regulatory functions. For example, upon deletion of segment (−9 to +36) of the p120ctn promoter there is still a noticeable promoter activity remained (Fig. 2b). This finding suggests the role of other cis-acting elements in the regulation of p120ctn promoter including the NFκB site immediately 5′ to the segment (−9 to +36).
Specificity protein 1 (SP1) belongs to a family of universally expressed zinc-finger DNA binding proteins that are involved in transcriptional regulation of many genes [26]. SP1 receives several signals from different intracellular pathways and multiple proteins including a number of other transcription factors that can interact with SP1 and thereby transduce their respective signal for gene regulation through SP1. Here, we describe the role of CRK (v-crk sarcoma virus CT10 oncogene homolog) in transcriptional down-regulation of p120ctn which are mediated through SP1.
CRK belongs to a family of widely expressed adaptor proteins that are involved in signal transduction from a variety of receptors and oncogenes. Examples include: Bcr-Abl; Tel-Abl, Erythropoietin receptor, EGFR, GM-CSF, Insulin receptor substrate, PDGF and VEGFR [1, 7]. Moreover, as a proto-oncogene, c-Crk expression has been linked to more aggressive types of tumors including aggressive forms of NSCLC. Measuring CRK mRNA by oligonucleotide array in 86 lung adenocarcinoma samples revealed that CRK mRNA was increased in more advanced, larger, and poorly differentiated tumors and correlated with shorter survival of patients [18]. Transcription of the CRK gene results in two different isoforms (CRK-I and CRK-II). These isoforms translate into 28 and 42 kDa proteins each one with diverse biological activities. Following stable transfection of CRK-I and CRK-II isoforms, all cell lines expressing CRK-I protein exhibited altered morphology, enhanced proliferation in soft agar, and heightened tumorigenesis in nude mice [17]. CRK-I/II interact with many downstream targets including: son of seven less homolog (SOS1); downstream of CRK (DOCK1) and stress activated protein kinase (JNK1). Although CRK-I and CRK-II are considered signaling adaptor proteins, they have both cytosolic and nuclear localizations [10, 16]. The nuclear localization of CRK suggests that this protein functions as a signal transmitter from the cytoplasm to the nucleus. Advancement toward understanding CRK biology was made through a screen for genes involved in cell-death in worms [22]. Through examination of mutated genes that were associated with lack of cell migration in C. elegans, a set of genes were identified the mammalian counterparts of which include Crk-II, Dock1, Rac1, and Elmo1. Lack of cell migration in this model was also observed in Crk-deficient worms, and this phenotype could be rescued by introduction of Rac1 or Dock1. As a result, it was suggested that Crk is positioned upstream of Rac1 in this pathway although direct targets downstream of Crk are still not well described. Our data shows that CRK negatively regulates p120ctn transcription in NSCLC cell lines through interaction with the SP1 transcription factor. It is worth mentioning that the effect of p120ctn on cell motility is also mediated through Rac1 [20]. The central role of CRK in signaling cascades makes it more likely that CRK affects several downstream targets, including SP1. Here we describe one of the mechanism through which CRK exerts its metastasis promoting role by down-regulation of p120ctn.
In summary, our data provide evidence for the role of proto-oncogene c-Crk in transcriptional down-regulation of p120ctn in NSCLC cells which is mediated through transcription factor SP1. Our data provides strong evidence in support of the interaction between SP1 and CRK-II. Considering SP1’s wide range of target genes and the central role of CRK in intracellular signaling, SP1-CRK interaction plays an important and central role in signal transduction.
These results along with our previous report on the role of FOXC2 in transcriptional down-regulation of p120ctn in NSCLC, identifies some of the metastasis promoting pathways that are abnormally activated in the process of tumorigenesis in NSCLC. Further attention into these pathways might provide additional insight to explain the aggressive behavior of these tumors.
References
Birge RB, Kalodimos C, Inagaki F, Tanaka S (2009) Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal 7:13
Boyd KE, Farnham PJ (1999) Coexamination of site-specific transcription factor binding and promoter activity in living cells. Mol Cell Biol 19:8393–8399
Bremnes RM, Veve R, Gabrielson E, Hirsch FR, Baron A, Bemis L, Gemmill RM, Drabkin HA, Franklin WA (2002) High-throughput tissue microarray analysis used to evaluate biology and prognostic significance of the E-cadherin pathway in non-small-cell lung cancer. J Clin Oncol 20:2417–2428
Chu S, Ferro TJ (2005) Sp1: regulation of gene expression by phosphorylation. Gene 348:1–11
Chung Y, Lam AK, Luk JM, Law S, Chan KW, Lee PY, Wong J (2007) Altered E-cadherin expression and p120 catenin localization in esophageal squamous cell carcinoma. Ann Surg Oncol 14:3260–3267
Davis MA, Ireton RC, Reynolds AB (2003) A core function for p120-catenin in cadherin turnover. J Cell Biol 163:525–534
Feller SM (2001) Crk family adaptors-signalling complex formation and biological roles. Oncogene 20:6348–6371
Grabe N (2002) AliBaba2: context specific identification of transcription factor binding sites. In Silico Biol 2:S1–S15
Ireton RC, Davis MA, van HJ, Mariner DJ, Barnes K, Thoreson MA, Anastasiadis PZ, Matrisian L, Bundy LM, Sealy L, Gilbert B, van RF, Reynolds AB (2002) A novel role for p120 catenin in E-cadherin function. J Cell Biol 159:465–476
Kar B, Reichman CT, Singh S, O’Connor JP, Birge RB (2007) Proapoptotic function of the nuclear Crk II adaptor protein. Biochemistry 46:10828–10840
Kelly KF, Spring CM, Otchere AA, Daniel JM (2004) NLS-dependent nuclear localization of p120ctn is necessary to relieve Kaiso-mediated transcriptional repression. J Cell Sci 117:2675–2686
Liu Y, Dong QZ, Zhao Y, Dong XJ, Miao Y, Dai SD, Yang ZQ, Zhang D, Wang Y, Li QC, Zhao C, Wang EH (2009) P120-catenin isoforms 1A and 3A differently affect invasion and proliferation of lung cancer cells. Exp Cell Res 315:890–898
Liu Y, Li QC, Miao Y, Xu HT, Dai SD, Wei Q, Dong QZ, Dong XJ, Zhao Y, Zhao C, Wang EH (2009) Ablation of p120-catenin enhances invasion and metastasis of human lung cancer cells. Cancer Sci 100:441–448
Liu Y, Wang Y, Zhang Y, Miao Y, Zhao Y, Zhang PX, Jiang GY, Zhang JY, Han Y, Lin XY, Yang LH, Li QC, Zhao C, Wang EH (2009) Abnormal expression of p120-catenin, E-cadherin, and small GTPases is significantly associated with malignant phenotype of human lung cancer. Lung Cancer 63:375–382
Liu Y, Xu HT, Dai SD, Wei Q, Yuan XM, Wang EH (2007) Reduction of p120(ctn) isoforms 1 and 3 is significantly associated with metastatic progression of human lung cancer. APMIS 115:848–856
Matsuda M, Nagata S, Tanaka S, Nagashima K, Kurata T (1993) Structural requirement of CRK SH2 region for binding to phosphotyrosine-containing proteins. Evidence from reactivity to monoclonal antibodies. J Biol Chem 268:4441–4446
Matsuda M, Tanaka S, Nagata S, Kojima A, Kurata T, Shibuya M (1992) Two species of human CRK cDNA encode proteins with distinct biological activities. Mol Cell Biol 12:3482–3489
Miller CT, Chen G, Gharib TG, Wang H, Thomas DG, Misek DE, Giordano TJ, Yee J, Orringer MB, Hanash SM, Beer DG (2003) Increased C-CRK proto-oncogene expression is associated with an aggressive phenotype in lung adenocarcinomas. Oncogene 22:7950–7957
Mortazavi F, An J, Dubinett S, Rettig M (2010) p120-catenin is transcriptionally downregulated by FOXC2 in non-small cell lung cancer cells. Mol Cancer Res 8:762–774
Noren NK, Liu BP, Burridge K, Kreft B (2000) p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol 150:567–580
Oas RG, Xiao K, Summers S, Wittich KB, Chiasson CM, Martin WD, Grossniklaus HE, Vincent PA, Reynolds AB, Kowalczyk AP (2010) p120-Catenin is required for mouse vascular development. Circ Res 106:941–951
Reddien PW, Horvitz HR (2000) CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat Cell Biol 2:131–136
Reynolds AB (2007) p120-catenin: Past and present. Biochim Biophys Acta 1773:2–7
Reynolds AB, Carnahan RH (2004) Regulation of cadherin stability and turnover by p120ctn: implications in disease and cancer. Semin Cell Dev Biol 15:657–663
Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103:843–852
Solomon SS, Majumdar G, Martinez-Hernandez A, Raghow R (2008) A critical role of Sp1 transcription factor in regulating gene expression in response to insulin and other hormones. Life Sci 83:305–312
Wang EH, Liu Y, Xu HT, Dai SD, Liu N, Xie CY, Yuan XM (2006) Abnormal expression and clinicopathologic significance of p120-catenin in lung cancer. Histol Histopathol 21:841–847
Wu C, Ma MH, Brown KR, Geisler M, Li L, Tzeng E, Jia CY, Jurisica I, Li SS (2007) Systematic identification of SH3 domain-mediated human protein-protein interactions by peptide array target screening. Proteomics 7:1775–1785
Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, Kowalczyk AP (2003) Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol 163:535–545
Xiao K, Oas RG, Chiasson CM, Kowalczyk AP (2007) Role of p120-catenin in cadherin trafficking. Biochim Biophys Acta 1773:8–16
Zhai B, Yan HX, Liu SQ, Chen L, Wu MC, Wang HY (2008) Reduced expression of P120 catenin in cholangiocarcinoma correlated with tumor clinicopathologic parameters. World J Gastroenterol 14:3739–3744
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Quantitative RT-PCRs showing the degree of mRNA expression following siRNA mediated silencing of p120ctn, CRK, AP2, SP1 and NF1 in A549 cells by two sets of siRNA for each gene. siRNAs marked with “*” were used in our experiments. (PPT 317 kb)
Supplemental Fig. 2
Western blots showing p120ctn protein level in H358 cells following expression of CRK-I, silencing SP1 or both. (PPT 207 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mortazavi, F., Dubinett, S. & Rettig, M. c-Crk proto-oncogene contributes to transcriptional repression of p120-catenin in non-small cell lung cancer cells. Clin Exp Metastasis 28, 391–404 (2011). https://doi.org/10.1007/s10585-011-9378-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-011-9378-8