
Abstract
Objectives
To test the hypothesis that more frequent enzyme replacement therapy (ERT) slows the decline in kidney function in adult patients with Fabry disease.
Methods
A single center open label 10-year prospective clinical trial of 12 patients with advanced Fabry disease who, after having experienced an ongoing decline in renal function after 2-4 years of receiving ERT at the approved dose of 0.2 mg/kg agalsidase alfa every other week (EOW), were switched to weekly (EW) ERT at the same dose. We used linear regression to fit each individual patient’s longitudinal estimated glomerular filtration rate (eGFR) record in order to compare the deterioration rates between EOW and EW ERT.
Results
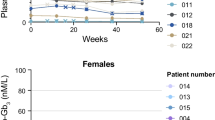
For the entire group, mean slope on agalsidase alfa every 2 weeks was -7.92 ± 2.88 ml/min/1.73 m2/year and 3.84 ± 4.08 ml/min/1.73 m2/year on weekly enzyme infusions (p = 0.01, two-tailed paired t test). Three patients (25 %) completed the entire study with relatively preserved renal function while 50 % of patients reached end-stage renal disease (ESRD) during the 10 years of this study. The estimated average delay to ESRD was 13.8 years [n = 11; 95 % CI 0.66, 27]. One patient had a positive eGFR slope on weekly infusions while the patient with the highest antibody titer had a steeper slope after switching. Mean globotriaosylceramide concentrations in urine and plasma as well as urine protein excretion remained unchanged.
Conclusions
Weekly enzyme infusions slow the decline of renal function in a subgroup of more severe patients thus showing that existing ERT can be further optimized.



Similar content being viewed by others


References
Altarescu G, Schiffmann R, Parker CC et al (2000) Comparative efficacy of dose regimens in enzyme replacement therapy of type I Gaucher disease. Blood Cells Mol Dis 26:285–290
Anderson LJ, Wyatt KM, Henley W et al (2014) Long-term effectiveness of enzyme replacement therapy in Fabry disease: results from the NCS-LSD cohort study. J Inherit Metab Dis 27(6):969–978. doi:10.1007/s10545-014-9717-4
Borgwardt L, Feldt-Rasmussen U, Rasmussen AK, Ballegaard M, Meldgaard Lund A (2013) Fabry disease in children: agalsidase-beta enzyme replacement therapy. Clin Genet 83:432–438
Brady RO, Schiffmann R (2000) Clinical features of and recent advances in therapy for Fabry disease. JAMA 284:2771–2775
Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L (1967) Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med 276:1163–1167
Branton MH, Schiffmann R, Sabnis SG et al (2002) Natural history of Fabry renal Disease: influence of alpha- galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore) 81:122–138
Coresh J, Turin TC, Matsushita K et al (2014) Decline in estimated glomerular filtration rate and subsequent risk of end-stage renal disease and mortality. JAMA 311:2518–2531
El Dib RP, Nascimento P, Pastores GM (2013) Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev 2, CD006663
EMA Fabrazyme: EPAR - Product Information In Editor ed.^eds. Book Fabrazyme: EPAR - Product Information. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000370/WC500020547.pdf. Accessed 27 July 2014
EMA Replagal: EPAR - Product Information. In Editor ed.^eds. Book Replagal: EPAR - Product Information. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000369/WC500053612.pdf. Accessed 24 July 2014
Eng CM, Banikazemi M, Gordon RE et al (2001) A phase 1/2 clinical trial of enzyme replacement in fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet 68:711–722
FDA FABRAZYME, BLA no. 103979 In Editor ed.^eds. Book FABRAZYME, BLA no. 103979. http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/103979s5135lbl.pdf. Accessed 24 July 2014
Hughes DA, Deegan PB, Milligan A et al (2013) A randomised, double-blind, placebo-controlled, crossover study to assess the efficacy and safety of three dosing schedules of agalsidase alfa enzyme replacement therapy for Fabry disease. Mol Genet Metab 109:269–275
Levey AS (2002) Clinical practice. Nondiabetic kidney disease. N Engl J Med 347:1505–1511
Lin HY, Huang YH, Liao HC et al (2014) Clinical observations on enzyme replacement therapy in patients with Fabry disease and the switch from agalsidase beta to agalsidase alfa. J Chin Med Assoc 77:190–197
Muenzer J, Wraith JE, Beck M et al (2006) A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med 8:465–473
Pisani A, Spinelli L, Visciano B et al (2013) Effects of switching from agalsidase Beta to agalsidase alfa in 10 patients with anderson-fabry disease. JIMD Rep 9:41–48
Ries M, Ramaswami U, Parini R et al (2003) The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr 162:767–772
Ries M, Gupta S, Moore DF et al (2005) Pediatric Fabry disease. Pediatrics 115:e344–e355
Ries M, Kim HJ, Zalewski CK et al (2007) Neuropathic and cerebrovascular correlates of hearing loss in Fabry disease. Brain 130:143–150
Rombach SM, Smid BE, Linthorst GE, Dijkgraaf MG, Hollak CE (2014) Natural course of Fabry disease and the effectiveness of enzyme replacement therapy: a systematic review and meta-analysis: effectiveness of ERT in different disease stages. J Inherit Metab Dis 37:341–352
Schiffmann R, Murray GJ, Treco D et al (2000) Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci U S A 97:365–370
Schiffmann R, Kopp JB, Austin HA 3rd et al (2001) Enzyme replacement therapy in fabry disease: a randomized controlled trial. JAMA 285:2743–2749
Schiffmann R, Floeter MK, Dambrosia JM et al (2003) Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve 28:703–710
Schiffmann R, Ries M, Timmons M, Flaherty JT, Brady RO (2006) Long-term therapy with agalsidase alfa for Fabry disease: safety and effects on renal function in a home infusion setting. Nephrol Dial Transplant 21:345–354
Schiffmann R, Askari H, Timmons M et al (2007) Weekly enzyme replacement therapy may slow decline of renal function in patients with Fabry disease who are on long-term biweekly dosing. J Am Soc Nephrol 18:1576–1583
Schiffmann R, Ries M, Blankenship D et al (2013) Changes in plasma and urine globotriaosylceramide levels do not predict Fabry disease progression over 1 year of agalsidase alfa. Genet Med 15:983–989
Sirrs SM, Bichet DG, Casey R et al (2014) Outcomes of patients treated through the Canadian Fabry disease initiative. Mol Genet Metab 111:499–506
Tondel C, Bostad L, Larsen KK et al (2013) Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol: JASN 24:137–148
Tsuboi K, Yamamoto H (2014) Clinical course of patients with Fabry disease who were switched from agalsidase-beta to agalsidase-alpha. Genet Med 16:766–772
van der Tol L, Smid BE, Poorthuis BJ et al (2014) A systematic review on screening for Fabry disease: prevalence of individuals with genetic variants of unknown significance. J Med Genet 51:1–9
Weidemann F, Niemann M, Stork S et al (2013) Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med 274:331–341
Weidemann F, Kramer J, Duning T et al (2014) Patients with Fabry disease after enzyme replacement therapy dose reduction versus treatment switch. J Am Soc Nephrol 25:837–849
Wraith JE, Tylki-Szymanska A, Guffon N et al (2008) Safety and efficacy of enzyme replacement therapy with agalsidase beta: an international, open-label study in pediatric patients with Fabry disease. J Pediatr 152:563–570, 570 e561
Acknowledgments
This work was supported in part by the Intramural Program of the National Institute of Neurological Disorders and Stroke, by Shire Human Genetic Therapies and the Baylor Healthcare System.
Conflict of interest
Markus Ries was an employee of Shire HGT from 2006 to 2009; he has served on advisory boards for Amicus, Alexion, GSK, and Shire HGT, has received consultancy honoraria from Alexion, Oxyrane, and Shire HGT as well as unrestricted research grants from Shire HGT in compliance with the policy of the Hospital of the University of Heidelberg, Germany. Raphael Schiffmann received research funds and honoraria from Shire HGT and Amicus Therapeutics.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Marc Patterson
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 147 kb)
Rights and permissions
About this article

Cite this article
Schiffmann, R., Swift, C., Wang, X. et al. A prospective 10-year study of individualized, intensified enzyme replacement therapy in advanced Fabry disease. J Inherit Metab Dis 38, 1129–1136 (2015). https://doi.org/10.1007/s10545-015-9845-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-015-9845-5