
Abstract
The aim of this study was to develop an analytical method to determine free concentrations of Europium (Eu(III)) in natural waters. Europium(III) in solution was detected using cathodic stripping voltammetry after complexation with N-nitroso-N-phenylhydroxylamine (cupferron). Optimization of analytical parameters allowed us to detect nanomolar levels of Eu(III) in solution. Free Eu(III) in solution was measured using the Donnan membrane technique in which a natural solution (the “donor”, containing various ligands) is separated from a ligand-free solution (the “acceptor”) by a cation-exchange membrane. This membrane allows only non-colloidal cationic species to pass through it, and after an adequate time equilibrium is reached between both compartments. Total Eu(III) concentration can then be quantified in the acceptor solution and related to free Eu(III) in the natural sample. Due to its high valency, free Eu(III) tends to adsorb strongly to the cation-exchange membrane. In order to determine the physicochemical conditions minimizing this adsorption, we analyzed solutions of different Eu(III) and Ca(II) (as background ion) concentrations. Results showed that 100 mM of Ca(II) were necessary to make adsorption of Eu(III) onto the membrane negligible. The optimized setup was then used to quantify Eu(III) complexation in a Eu(III)-dissolved organic matter solution.
Similar content being viewed by others

1 Introduction
In recent years, the growing utilization of rare earth elements (REEs) in high end technologies led to increasing levels of REEs in natural waters, usually in the ng/L range (Bau et al. 2018; Gwenzi et al. 2018; Hatje et al. 2016; Klaver et al. 2014; Lerat-Hardy et al. 2019; Liu et al. 2019). Nonetheless, toxicological and environmental impact of these elements is mostly unknown (Gonzalez et al. 2014; Mestre et al. 2019; Romero-Freire et al. 2019). The potential toxicity of a metallic contaminant is linked to its elemental speciation and particularly to the free ion species in solution. Thus, it is necessary to develop analytical speciation techniques with very low detection limits. The quantitative speciation can then be used to calibrate thermodynamic models, crucial tools for predicting the environmental fate of trace metals. Currently, calibration of thermodynamic models for REEs binding to natural ligands (natural organic matter and mineral surfaces) has been performed on rather limited datasets and obtained generally from synthetic systems or using purified components (Janot et al. 2013; Marsac et al. 2010, 2021; Milne et al. 2003). Thus, their applicability to natural systems still needs to be confirmed. This work aims to develop an analytical setup capable to measure Eu speciation at nanomolar level, by combining the use of field DMT cells and portable electrochemical devices (Parat and Pinheiro 2015; Weng et al. 2011). This approach serves a first step to test the applicability of the REEs thermodynamic models to natural systems.
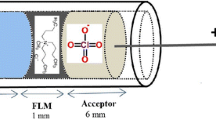
Among available speciation techniques, the Donnan membrane technique (DMT) is a sensitive and efficient method for quantifying free trace metals in solution. In this method, the sample solution (“donor”) is separated from an “acceptor” solution by a cation-exchange membrane. This membrane, made of polystyrene and divinylbenzene and charged with sulfonic acid groups is fully deprotonated at pH > 2, allowing only non-colloidal cationic species to pass through it (Weng et al. 2011, 2002). At Donnan equilibrium (48 h for divalent cations), the ionic strength is equal on both sides of the membrane and activity ratios of free cations in the donor and the acceptor solution are equal: since complexed species are not able to pass through the membrane, the total concentration of metal in the acceptor side corresponds to the free concentration in the donor side. This technique has been efficiently used for the determination of free concentration of divalent metals (Cd2+, Cu2+, Ni2+, Pb2+, Zn2+), both in the lab (Marang et al. 2006; Parat and Pinheiro 2015; Zelano et al. 2018) and in various natural samples such as surface waters (Kalis et al. 2006; Sigg et al. 2006), soil solutions (Cancès et al. 2003; Ren et al. 2015; Weng et al. 2001) or even manure slurry (van der Stelt et al. 2005) and wine (Lao et al. 2018). Conversely, the application to trivalent metals such as REEs has not been largely tested. Weng et al. measured free Al3+ concentration in gibbsite suspensions and soil solutions and found a longer equilibration time than for divalent cations (3–4 days) (Weng et al. 2002). Jones et al. have analyzed Al, Fe and REEs in drainage waters from coastal sediments using DMT and found an equilibration time of 50 h (Jones et al. 2016). In the DMT setup, the presence of a multivalent cation in the background solution (usually, Ca2+) is necessary to compete with the target metal for the binding to the membrane and to ensure sufficient transport of the target cation through the membrane. The concentration of Ca2+ in the electrolyte has been shown to have a great influence on the transport of trivalent metals through the DMT membrane (Weng et al. 2002), yet no study investigating free trivalent metal pool determined the impact of salinity (as Ca2+ level) on the experimental results (Jones et al. 2016; Weng et al. 2002).
Detection of REEs with electrochemistry has been developed using different techniques (square wave, differential pulse, cathodic stripping) and different working electrodes (glassy carbon rotating disk and mercury drop electrodes) (Abollino et al. 1997; Grabarczyk and Wardak 2014; Janos and Synek 2005; Moretto et al. 2000; Schroll et al. 2013; Schumacher et al. 2010; Ugo et al. 1990; Yantasee et al. 2006; Zelic 2003). In this work, we used cathodic stripping voltammetry (CSV) which allows a very low detection limit and robust measurements (Abollino et al. 1997; Grabarczyk and Wardak 2014). In this technique, the metal of interest (europium) is complexed by a specific ligand and adsorbs at the electrode, before applying a potential scan toward negative values. The resulting reductive current is proportional to the metal concentration. Calibration of this method in various metal and salinity media is needed to determine the detection limit and applicability of this analytical approach.
The objective of this study is to determine the free fraction of Eu3+ in solution under various relevant metal and calcium concentration conditions. Thus, we determined the experimental conditions under which Eu concentrations can be measured in the acceptor side of a DMT setup, in particular the influence of salt concentration on both Eu transport through the membrane and electrochemical signal. After calibration, this method was validated by quantifying free Eu3+ in a solution containing dissolved organic matter (DOM) isolated from groundwater.
2 Materials and Methods
2.1 Reagents
All reagents were prepared using ultrapure water (18.2 MΩ cm−1). Europium standard solution used was certified reference material (1002 mg L−1). Cupferron (N-nitroso-N-phenylhydroxylamine) was supplied by Sigma-Aldrich. Nitric acid was of supra-pure grade. Ca(NO3)2 was of analytical grade.
Cation-exchange membranes were purchased from VWR (Ref. 551652U) (Temminghoff et al. 2000). Before each utilization, membranes were washed to remove any undesired cations from their surface (Kalis et al. 2006). In order to do so, membranes were immersed for at least 30 min in the following solutions: 1 M ethylenediaminetetraacetic acid (EDTA), ultrapure water, 0.1 M HNO3, ultrapure water, 1 M Ca(NO3)2 (to saturate membrane surfaces with Ca2+). Each step was repeated twice. Finally, membranes were conditioned in a Ca(NO3)2 solution of targeted background concentration.
2.2 Electrochemical Detection by Square Wave Cathodic Stripping Voltammetry
Voltammetric scans were obtained with an EcoChemie μAutolab III and a PGStat 12 in conjunction with a Metrohm 663 VA stand. A three-electrode configuration was used, comprising a hanging drop mercury electrode (HDME) as a working electrode (drop size: 2), a Ag/AgCl reference electrode and a Pt counter electrode. The setup was controlled by GPES 4.9 software.
Acetate buffer and cupferron were added to the 20 ml sample solution in a voltammetric cell. The cell was connected to the electrode stand, and the solution was N2-purged until signal was stable, at least 600 s. Pre-concentration potential was set to − 0.005 V for a 60 s accumulation time. After 10 s of equilibration, stirring was stopped, and potential scan from − 0.5 to − 1 V was recorded at a 25 Hz frequency (Abollino et al. 1997). After recording the blank signal, additions of known amount of Eu solution were performed, and procedure was repeated. Blank scans were subtracted from the sample scans before peak analysis.
2.3 Eu(III) Adsorption Onto Cation-Exchange Membrane
Preliminary results had shown that no Eu(III) was going through the DMT membrane during an equilibration experiment, even in the presence of ligand in the acceptor side (data not shown), probably due to strong accumulation of Eu(III) within the negatively charged membrane at the low Ca level used (2 mM). To test this hypothesis and determine the impact of counter-ion concentration on Eu accumulation, we placed 1 cm2 of DMT membrane in 50-mL solutions of 0.1, 1 and 5 µM Eu(III) at different Ca levels (2.5, 10 and 100 mM Ca(NO3)2). After 4 days of equilibration, Eu(III) concentration left in solution was measured by CSV.
2.4 Eu(III) Speciation Using the Donnan Membrane Technique
The DMT setup was similar to the one described in Zelano et al. (2018). The acceptor solution was placed inside a cylindrical poly(methyl methacrylate) (PMMA) cell filled with 10 mL of the Ca(NO3)2 electrolyte solution and directly immersed in a 320 ml-donor solution of the same electrolyte composition from which it was separated by two cation-exchange membranes. PMMA cells were washed twice in 5% HNO3 and rinsed with ultrapure water. Membranes were washed as described in the previous paragraph. Five setups were installed containing 20 mg/L of natural DOM and various initial Eu concentrations (0.5, 1, 3, 5 and 10 µM). This DOM was obtained from Wageningen University and originates from groundwater in the Netherlands. It is mainly (> 80%) composed of fulvic acids (FA), as determined by the method of van Zomeren and Comans (van Zomeren and Comans 2007). The experiment was run subsequently at three increasing pH values (3.9, 4.6 and 5.2). pH was measured at the beginning and the end of the equilibration time, and only the final value was considered for the interpretation. An equilibration time of 4 days was used based on published experiments using trivalent metals with DMT (Jones et al. 2016; Weng et al. 2002). When acceptor compartments were emptied and sampled for one pH condition, they were immediately filled with blank solution of the next target pH value and reimmersed in the donor solution adjusted at the same pH value.
2.5 Speciation Prediction with NICA-Donnan Modeling
Eu(III) speciation in solution in the presence of groundwater DOM was predicted using the ORCHESTRA software, 2020 version (Meeussen 2003). The solutions were assumed to be in equilibrium with atmospheric CO2 (pCO2 = 3.9E−4 atm.). We used the standard Minteq-V4 thermodynamic database provided in ORCHESTRA to which we added the thermodynamic constants of lanthanide species according to NIST (Martell et al. 1995) and the hydroxy species Ln-OH2+, Ln-(OH)2+ and Ln-(OH)3 from Lee and Byrne (Lee and Byrne 1992). Similar to Janssen and Verweij (Janssen and Verweij 2003), we used additional thermodynamic constants for Ln-HCO32+ from Millero (Millero 1992) and for Ln-CO3+ and Ln-(CO3)2− from Lee and Byrne (Lee and Byrne 1993). For speciation calculation, groundwater DOM was considered to be 82% reactive (as FA only), and complexation of Eu to FA was calculated with the NICA (non-ideal competitive adsorption)-Donnan model (Kinniburgh et al. 1996; Koopal et al. 2005) using the generic proton, calcium and Eu binding parameters derived by Milne et al. (Milne et al. 2003). Activity coefficients were calculated using the Davies equation.
At the high Ca concentration used in the DMT experiment (0.1 M), we considered the difference of Ca activities between both compartments to be negligible and no ionic strength correction was applied. Under these conditions, the resulting high NO3− concentration (200 mM) leads to a significant amount of Eu(NO3)2+ in solution. Thus, the pool of Eu equilibrated in the acceptor side of the DMT field cell is composed of the inorganic fraction of the whole Eu pool, that is both free Eu3+ and Eu(NO3)2+. The proportion of carbonate Eu species was calculated using ORCHESTRA and found negligible compared to the two main inorganic Eu species (see Figure S1 from the Supplementary Information).
3 Results
3.1 Detection of Eu with Cathodic Stripping Voltammetry
3.1.1 Optimization of Analytical Method
To achieve the best analytical performance, the effect of chemical and instrumental parameters known to have a significant influence on the peak current was examined. Optimized values determined in a previous study (Abollino et al. 1997) were confirmed here: 10–2 M acetate buffer and 6·10–5 M cupferron concentrations were used, deposition potential and time were fixed at − 0.005 V and 60 s, respectively. The pH was adjusted to 5.5 with HNO3, an optimal value between protonation of the cupferron and hydrolysis of the europium (7.2). Under these conditions, the presence of europium gives a well-developed peak, with a maximum potential around − 0.75 V (see Figure S2a from the Supplementary Information).
To determine the impact of Ca concentration on Eu equilibration with DMT setup, we first had to verify its influence on CSV signal. Figure 1 shows that when Ca(NO3)2 concentration increases, a large peak centered around − 0.81 V appears, at a potential close to the peak potential of europium (detailed in Fig. 1a). Ca(NO3)2 used was of analytical grade (≥ 99%) but can still contain impurities such as trace metals (< 5 ppm according to the notice) that can induce current in the voltammetric cell, noticeable at large concentration such as the one used here.

a Influence of calcium concentration on electroanalytical signal and b background subtracted voltammogram used for calibration in 2.5 mM Ca (Eu concentrations listed in the Figure)
To get rid of this background signal, a corresponding blank voltammogram was recorded at the beginning of each day and subtracted from all the following measures. Increase in Eu peak around 0.75 V is better evidenced in background subtracted voltammograms, as shown in Fig. 1b. Data processing and peak height determination were conducted using R software version 3.5.3 (R Core Team 2019) and R packages spectacles (Roudier 2019) and chemCal version 0.2.1 (https://CRAN.R-project.org/package=chemCal). Details and example of R script are given in supplemental material.
3.1.2 Calibration Curves
To quantify the linearity and the sensitivity of the method, we determined calibration curves at different Ca concentration. For each Eu concentration, CSV signals were recorded in triplicate. The three calibration curves up to 150, 100 and 50 nM Eu at 2.5, 10 and 100 mM Ca, respectively, are shown in Fig. 2, and corresponding electrochemical signals are shown in supplementary information (Figures S3–S5). The CSV response is linear at low metal concentration, as expected, and the slope of the linear regression increases with Ca concentration. However, when metal concentration increases (above 80 nM Eu in 2.5 mM Ca medium), peak height levels off due to electrode surface saturation (Abollino et al. 1997). Above this concentration, when needed, calibration curves can be fitted using quadratic equation (see Figure S3 for calibration curve at 2.5 mM Ca). Limits of detection were 2–5 nM depending on the calibration curve.

Calibration curve of europium at 2.5, 10 and 100 mM Ca
3.2 Accumulation of Eu Within the Cation-Exchange Membrane
To determine Eu accumulation within the cation-exchange membrane, 1 cm2 of membrane was equilibrated in 50 ml of Eu solution. After 4 days of contact, an aliquot of solution was diluted in an acetate-buffered solution at 2.5 or 10 mM Ca(NO3)2, and Eu concentration in the solution was quantified using standard addition method. All CSV signals were recorded in triplicate. For high Eu concentration (final concentration > 60 nM), results were fitted with both linear and quadratic equations. However, both values were contained within the experimental error, so only linear fitting results were kept (Figure S6). Results depending on Eu and Ca concentrations are given in Table 1 and Fig. 3. Example of standard addition data processing and fitting is given in supporting information.

Eu accumulation within the cation-exchange membrane depending on initial Eu and Ca concentrations in the solution
Due to its higher valency, we observe a preferential accumulation of Eu3+ within the negatively charged membrane, as previously evidenced for Al3+ (Weng et al. 2002). At freshwater representative Ca concentration (2.5 mM), nearly all Eu3+ are accumulated within the membrane and disappeared from the solution. Our results evidence that a large excess of background cation (100 mM Ca) is necessary to prevent Eu(III) accumulation in the membrane, demonstrating the strong affinity of Eu(III) toward these cationic-exchange membranes.
Error propagation due to dilution of the initial sample in the solution for CSV measurements leads to high uncertainty for some samples, especially at low initial concentration. Data processing for determining the Eu peak height in the CSV signal has also large impact on the calculated Eu concentration, so we propose an automatic procedure to determine the peak limits in order to reduce subjectivity (see Supplementary material for example of R script).
3.3 Determination of Free Eu3+ in Presence of DOM
We used the optimal electroanalytical and chemical (100 mM Ca concentration) parameters determined previously to quantify Eu binding to DOM using the field DMT setup (Kalis et al. 2006; van der Stelt et al. 2005). After 4d equilibration, the acceptor solution was sampled, and Eu concentration was determined by CSV. Eu concentration in the voltammetric cell was determined by the standard addition method as described in the previous paragraph, after diluting an aliquot in a 10 mM Ca solution. Linear regression results showed that with this approach, Eu concentration in the acceptor compartment is determined with a confidence interval of 10%. Results show an increase in Eu binding to organic matter with increasing pH, as expected (Janot et al. 2011) (see Fig. 4). Below pH 4, almost no Eu is bound by the DOM, and 40–70% is complexed to the DOM at pH > 5 (Figure S7).

Eu binding to groundwater DOM depending on pH and Eu concentration. Symbols are experimental data with confidence limits, lines are NICA-Donnan predictions with generic proton and Eu binding parameters for fulvic acids; [Ca] = 100 mM, [FA] = 16.4 mg/L
4 Discussion
4.1 Applicability to Field Measurements
The aim of this study was to develop an in situ method for determining free Eu concentrations in natural samples. By coupling CSV measurements to a field DMT setup, we managed to quantify free Eu in solution in the presence of dissolved organic matter with an excellent detection limit (~ 3 nM). However, the high affinity of cation-exchange membrane for trivalent Eu makes it necessary to work using a high concentration of counter-ion Ca (100 mM), thus limiting the applicability of the method to saline environments.
Calibration curves showed that the detection limits of our electroanalytical method are around 3 nM Eu. This is still above Eu background concentration in natural waters (Bau et al. 2018; Hatje et al. 2016), but this technique can be used to detect concerning levels of Eu, for example, in mining-impacted or contaminated environments (Liu et al. 2019). We thus demonstrate that this analytical technique competes well with the classical ones used for europium quantification, such as inductively coupled plasma-atomic emission spectroscopy (ICP-AES) or even time-resolved laser spectroscopy (TRLS) (Berthoud et al. 1989; Janot et al. 2011; Maind et al. 2008), but has a detection limit still 1 to 2 orders of magnitude higher than the one of ICP-mass spectrometry (ICP-MS) (Hatje et al. 2016; Kautenburger and Beck 2010; Leguay et al. 2016). However, ICP techniques are very sensitive to matrix effects, especially high salt concentrations needed by the DMT speciation setup. TRLS is a very efficient technique to detect lanthanides in solution, and allows their quantification by standard additions into a 3 M K2CO3 medium (Berthoud et al. 1989; Janot et al. 2011), however, it requires a very specific setup. Compared to these techniques used in the literature, great advantages of electrochemical detection are its low cost and its portability, which could be even improved by cell miniaturization.
Among electroanalytical techniques, several studies using Nafion-coated glassy carbon electrode reported comparable low detection limits, on the order of 10 nM or even down to 10−11 M (Moretto et al. 2000). We first tested this setup and procedure and indeed obtained good detection limits. However, the reproducibility of the Nafion coating was extremely poor, and we advise against the use of this method. In our trials, we had no success determining low Eu concentration using other electrodes than the hanging mercury drop electrode (HMDE), thus limiting the miniaturization of the analytical device. Nevertheless, we were successful in designing a reproducible method to quantitatively determine low levels of Eu in solution under various salinity conditions, by using using CSV and the HMDE.
One limitation of the experimental setup we propose is the high concentration of background ion (Ca2+) needed to prevent accumulation of the trivalent Eu within the cation-exchange membrane, leading to analytical matrixes that are not representative of most environmental conditions. It will indeed be impossible to install the DMT field cell in freshwaters: in these conditions, all trace trivalent metals will accumulate within the membranes and not in the acceptor compartment. However, the whole setup can be used in high salinity environments such as brine pore water of host rocks for high-level nuclear waste (Banik et al. 2017; Marsac et al. 2017; Schnurr et al. 2015) or salt marshes close to marine waters or coastal waters (Jones et al. 2016).
Indeed, Jones et al. also used a cation-exchange membrane to determine the free amount of REEs in natural acidic sulfate soil drainage waters (Jones et al. 2016). These authors observed a very small amount of REEs in the acceptor compartment and concluded that a large proportion of REEs was present as negatively charged REEs species (DOM-complexes and disulfate species). However, the maximal Ca concentration in their samples was 4 mM, thus from the results of our study we can expect that a large part of the REEs was actually accumulated within the membrane in their measurements.
4.2 Contribution to the Improvement of Model Predictions
Using our setup combining electrochemical detection and field DMT cell, we were able to quantify Eu binding to a groundwater DOM at various pH values in a 100 mM Ca solution. This information is crucial to determine relevant metal-binding parameters for natural organic matter, using ion-binding models such as NICA-Donnan (Kinniburgh et al. 1996; Koopal et al. 2005; Milne et al. 2003). The use of such thermodynamic models that can predict rare earth elements partitioning and speciation in terrestrial and aquatic systems is of key importance for risk assessment; however, most of the rare earth binding parameters are still lacking.
Experimental results for measured free Eu in Eu-DOM solution were compared with free inorganic europium amount (i.e., Eu3+ and Eu(NO3)2+) predicted by the NICA-Donnan model. At pH < 4, there is nearly any binding to DOM and there is rather good agreement between the measured and calculated free Eu3+ concentrations (Fig. 5). At higher pH (around 4.6 and 5.2), the model substantially underestimates the Eu binding to the organic matter and predicts more free inorganic Eu in solution than what was actually observed. Leguay et al. (2016) also observed discrepancies between modeling predictions (using NICA-Donnan, but also WHAM VII and Stockholm Humic Model) and experimental results of Eu binding to humic acid in 100 mM NaNO3 (as detected by ion-exchange technique) at pH > 4 (Leguay et al. 2016). Interestingly, our study and the study of Leguay et al. (2016) show opposite results using two different DOM proxies, a groundwater fulvic acid in our case and a surface water humic acid (Suwannee River Standard II) in their case. This illustrates once more the limitation of applying published generic binding parameters for humic substances (Botero et al. 2017; Jones et al. 2016). It is necessary to remember that these generic parameters, when derived from sufficient data, represent only an average behavior of humic substances, but differences between them give rise to uncertainties in the predicted speciation (Groenenberg et al. 2010). Here, what the results of this modeling show that our experimental results are within the same range of the previous studies used to derive the generic binding parameters. This method can thus be used to obtain new data sets needed to derive better metal-binding parameters for various humic substances.

Comparison between measured and calculated free Eu concentrations (Eu3+ and EuNO32+) in the presence of 16.4 mgFA/L and 100 mM Ca(NO3)2, [Eu]total = 0.5–10 µM. Dashed line represents the 1:1 agreement between measurements and model prediction
5 Conclusions
The objective of this study was to determine the applicability of linking electrochemical detection to Donnan membrane speciation technique to determine free Eu concentration under environmentally relevant conditions. The results show a very good reproducibility and detection limit of cathodic stripping voltammetry using a hanging-drop mercury electrode for low levels of Eu in solution. However, due to the strong interactions between the cation-exchange membrane and the europium, the implementation of the DMT was more difficult for the trivalent REE than for divalent metals ions (Cu, Zn, Cd, Ni). It was determined that a high concentration of counter-ion Ca2+ (0.1 M) was needed to prevent accumulation of Eu3+ within the negatively charged membrane, thus making this technique ideal for high salinity environments such as estuaries waters or pore waters of nuclear waste rock hosts.
6 Supporting Information
Raw and processed data for calibration curves, standard addition curves and an example of data processing code (R markdown) can be found in Supplementary material.
References
Abollino O, Aceto M, Mentasti E, Sarzanini C, van den Berg CMG (1997) Determination of trace europium by adsorptive cathodic stripping voltammetry after complexation with cupferron. Electroanalysis 9:444–448. https://doi.org/10.1002/elan.1140090603
Banik NL, Marsac R, Lützenkirchen J, Marquardt CM, Dardenne K, Rothe J, Bender K, Geckeis H (2017) Neptunium sorption and redox speciation at the illite surface under highly saline conditions. Geochim Cosmochim Acta 215:421–431. https://doi.org/10.1016/j.gca.2017.08.008
Bau M, Schmidt K, Pack A, Bendel V, Kraemer D (2018) The European Shale: An improved data set for normalisation of rare earth element and yttrium concentrations in environmental and biological samples from Europe. Appl Geochem 90:142–149. https://doi.org/10.1016/j.apgeochem.2018.01.008
Berthoud T, Decambox P, Kirsch B, Mauchien P, Moulin C (1989) Direct determination of traces of lanthanide ions in aqueous solutions by laser-induced time-resolved spectrofluorimetry. Anal Chim Acta 220:235–241. https://doi.org/10.1016/S0003-2670(00)80266-4
Botero WG, Pineau M, Janot N, Domingos RF, Mariano J, Rocha LS, Groenenberg JE, Benedetti MF, Pinheiro JP (2017) Isolation and purification treatments change the metal-binding properties of humic acids: effect of HF/HCl treatment. Environ Chem 14:417. https://doi.org/10.1071/EN17129
Cancès B, Ponthieu M, Castrec-Rouelle M, Aubry E, Benedetti MF (2003) Metal ions speciation in a soil and its solution: experimental data and model results. Geoderma 113:341–355. https://doi.org/10.1016/S0016-7061(02)00369-5
Gonzalez V, Vignati DAL, Leyval C, Giamberini L (2014) Environmental fate and ecotoxicity of lanthanides: Are they a uniform group beyond chemistry? Environ Int 71:148–157. https://doi.org/10.1016/j.envint.2014.06.019
Grabarczyk M, Wardak C (2014) A new voltammetric strategy for sensitive and selective determination of gallium using cupferron as a complexing agent. J Environ Sci Health Part A 49:1142–1148. https://doi.org/10.1080/10934529.2014.897156
Groenenberg JE, Koopmans GF, Comans RNJ (2010) Uncertainty analysis of the nonideal competitive adsorption−donnan model: effects of dissolved organic matter variability on predicted metal speciation in soil solution. Environ Sci Technol 44:1340–1346. https://doi.org/10.1021/es902615w
Gwenzi W, Mangori L, Danha C, Chaukura N, Dunjana N, Sanganyado E (2018) Sources, behaviour, and environmental and human health risks of high-technology rare earth elements as emerging contaminants. Sci Total Environ 636:299–313. https://doi.org/10.1016/j.scitotenv.2018.04.235
Hatje V, Bruland KW, Flegal AR (2016) Increases in anthropogenic gadolinium anomalies and rare earth element concentrations in san francisco bay over a 20 year record. Environ Sci Technol 50:4159–4168. https://doi.org/10.1021/acs.est.5b04322
Janos P, Synek V (2005) Differential pulse polarographic determination of europium in the presence of humic substances. Talanta 66:188–193. https://doi.org/10.1016/j.talanta.2004.11.008
Janot N, Benedetti MF, Reiller PE (2011) Colloidal α-Al2O3, europium(III) and humic substances interactions: a macroscopic and spectroscopic study. Environ Sci Technol 45:3224–3230. https://doi.org/10.1021/es102592a
Janot N, Reiller PE, Benedetti MF (2013) Modelling Eu(III) speciation in a Eu(III)/PAHA/α-Al2O3 ternary system. Colloids Surf Physicochem Eng Asp 435:9–15. https://doi.org/10.1016/j.colsurfa.2013.02.052
Janssen RPT, Verweij W (2003) Geochemistry of some rare earth elements in groundwater, Vierlingsbeek. The Netherlands Water Res 37:1320–1350. https://doi.org/10.1016/S0043-1354(02)00492-X
Jones AM, Xue Y, Kinsela AS, Wilcken KM, Collins RN (2016) Donnan membrane speciation of Al, Fe, trace metals and REEs in coastal lowland acid sulfate soil-impacted drainage waters. Sci Total Environ 547:104–113. https://doi.org/10.1016/j.scitotenv.2016.01.024
Kalis EJJ, Weng D, F., Temminghoff, E.J.M., Van Riemsdijk, W.H., (2006) Measuring free metal ion concentrations in situ in natural waters using the donnan membrane technique. Environ Sci Technol 40:955–961. https://doi.org/10.1021/es051435v
Kautenburger R, Beck HP (2010) Influence of geochemical parameters on the sorption and desorption behaviour of europium and gadolinium onto kaolinite. J Environ Monit 12:1295–1301. https://doi.org/10.1039/b914861b
Kinniburgh DG, Milne CJ, Benedetti MF, Pinheiro JP, Filius J, Koopal LK, Van Riemsdijk WH (1996) Metal Ion binding by humic acid: application of the NICA-Donnan model. Environ Sci Technol 30:1687–1698. https://doi.org/10.1021/es950695h
Klaver G, Verheul M, Bakker I, Petelet-Giraud E, Négrel P (2014) Anthropogenic rare earth element in rivers: gadolinium and lanthanum. Partitioning between the dissolved and particulate phases in the Rhine River and spatial propagation through the Rhine-Meuse Delta (The Netherlands). Appl Geochem 47:186–197. https://doi.org/10.1016/j.apgeochem.2014.05.020
Koopal LK, Saito T, Pinheiro JP, van Riemsdijk WH (2005) Ion binding to natural organic matter: general considerations and the NICA–Donnan model. Colloids Surf Physicochem Eng Asp 265:40–54. https://doi.org/10.1016/j.colsurfa.2004.11.050
Lao M, Companys E, Weng L, Puy J, Galceran J (2018) Speciation of Zn, Fe, Ca and Mg in wine with the Donnan membrane technique. Food Chem 239:1143–1150. https://doi.org/10.1016/j.foodchem.2017.07.040
Lee JH, Byrne RH (1993) Complexation of trivalent rare earth elements (Ce, Eu, Gd, Tb, Yb) by carbonate ions. Geochim Cosmochim Acta 57:295–302. https://doi.org/10.1016/0016-7037(93)90432-V
Lee JH, Byrne RH (1992) Extinction of comparative rare earth element complexation behavior using linear free-energy relationships. Geochim Cosmochim Acta 56:1127–1137. https://doi.org/10.1016/0016-7037(92)90050-S
Leguay S, Campbell PGC, Fortin C (2016) Determination of the free-ion concentration of rare earth elements by an ion-exchange technique: implementation, evaluation and limits. Environ Chem 13:478. https://doi.org/10.1071/EN15136
Lerat-Hardy A, Coynel A, Dutruch L, Pereto C, Bossy C, Gil-Diaz T, Capdeville M-J, Blanc G, Schäfer J (2019) Rare earth element fluxes over 15 years into a major european estuary (Garonne-Gironde, SW France): hospital effluents as a source of increasing gadolinium anomalies. Sci Total Environ 656:409–420. https://doi.org/10.1016/j.scitotenv.2018.11.343
Liu W-S, Guo M-N, Liu C, Yuan M, Chen X-T, Huot H, Zhao C-M, Tang Y-T, Morel JL, Qiu R-L (2019) Water, sediment and agricultural soil contamination from an ion-adsorption rare earth mining area. Chemosphere 216:75–83. https://doi.org/10.1016/j.chemosphere.2018.10.109
Maind SD, Chattopadhyay N, Gandhi Ch, Kumar SC, Sudersanan M (2008) Quantitative evaluation of europium in blue ballpoint pen inks/offset printing inks tagged with europium thenoyltrifluoroacetonate by spectrofluorometry and ICP-AES. Sci Justice 48:61–66. https://doi.org/10.1016/j.scijus.2007.09.007
Marang L, Reiller P, Pepe M, Benedetti MF (2006) Donnan Membrane Approach: From Equilibrium to Dynamic Speciation. Environ Sci Technol 40:5496–5501. https://doi.org/10.1021/es060608t
Marsac R, Banik NL, Lützenkirchen J, Catrouillet C, Marquardt CM, Johannesson KH (2017) Modeling metal ion-humic substances complexation in highly saline conditions. Appl Geochem 79:52–64. https://doi.org/10.1016/j.apgeochem.2017.02.004
Marsac R, Catrouillet C, Davranche M, Bouhnik-Le Coz M, Briant N, Janot N, Otero-Fariña A, Groenenberg JE, Pédrot M, Dia A (2021) Modeling rare earth elements binding to humic acids with model VII. Chem Geol 567:120099
Marsac R, Davranche M, Gruau G, Dia A (2010) Metal loading effect on rare earth element binding to humic acid: Experimental and modelling evidence. Geochim Cosmochim Acta 74:1749–1761. https://doi.org/10.1016/j.gca.2009.12.006
Martell AE, Smith RM, Motekaitis RJ (1995) Critical stability constants of metal complexes database (NIST standard reference database 46 No. Version 2). US department of Commerce, Gaithersburg
Meeussen JCL (2003) ORCHESTRA: an object-oriented framework for implementing chemical equilibrium models. Environ Sci Technol 37:1175–1182. https://doi.org/10.1021/es025597s
Mestre NC, Sousa VS, Rocha TL, Bebianno MJ (2019) Ecotoxicity of rare earths in the marine mussel Mytilus galloprovincialis and a preliminary approach to assess environmental risk. Ecotoxicology 28:294–301. https://doi.org/10.1007/s10646-019-02022-4
Millero FJ (1992) Stability constants for the formation of rare earth-inorganic complexes as a function of ionic strength. Geochim Cosmochim Acta 56:3123–3132. https://doi.org/10.1016/0016-7037(92)90293-R
Milne CJ, Kinniburgh DG, van Riemsdijk WH, Tipping E (2003) Generic NICA−Donnan model parameters for metal-ion binding by humic substances. Environ Sci Technol 37:958–971. https://doi.org/10.1021/es0258879
Moretto LM, Brunetti B, Chevalet J, Ugo P (2000) Multiple square wave voltammetry of nanomolar and subnanomolar concentrations of europium(III) at polymer-coated electrodes. Commun, Electrochem. https://doi.org/10.1016/S1388-2481(99)00170-8
Parat C, Pinheiro JP (2015) ISIDORE, a probe for in situ trace metal speciation based on Donnan membrane technique with related electrochemical detection part 1: Equilibrium measurements. Anal Chim Acta 896:1–10. https://doi.org/10.1016/j.aca.2015.07.016
Ren Z-L, Tella M, Bravin MN, Comans RNJ, Dai J, Garnier J-M, Sivry Y, Doelsch E, Straathof A, Benedetti MF (2015) Effect of dissolved organic matter composition on metal speciation in soil solutions. Chem Geol 398:61–69. https://doi.org/10.1016/j.chemgeo.2015.01.020
Romero-Freire A, Joonas E, Muna M, Cossu-Leguille C, Vignati DAL, Giamberini L (2019) Assessment of the toxic effects of mixtures of three lanthanides (Ce, Gd, Lu) to aquatic biota. Sci Total Environ 661:276–284. https://doi.org/10.1016/j.scitotenv.2019.01.155
Roudier P (2019) Spectacles: storing and manipulating spectroscopy data in R
Schnurr A, Marsac R, Rabung T, Lützenkirchen J, Geckeis H (2015) Sorption of Cm(III) and Eu(III) onto clay minerals under saline conditions: batch adsorption, laser-fluorescence spectroscopy and modeling. Geochim Cosmochim Acta 151:192–202. https://doi.org/10.1016/j.gca.2014.11.011
Schroll CA, Chatterjee S, Levitskaia TG, Heineman WR, Bryan SA (2013) Electrochemistry and Spectroelectrochemistry of Europium(III) Chloride in 3LiCl–2KCl from 643 to 1123 K. Anal Chem 85:9924–9931. https://doi.org/10.1021/ac402518p
Schumacher PD, Woods N, a., Schenk, J.O., Clark, S.B., (2010) Preconcentration of trivalent lanthanide elements on a mercury film from aqueous solution using rotating disk electrode voltammetry. Anal Chem 82:5663–5668. https://doi.org/10.1021/ac101180w
Sigg L, Black F, Buffle J, Cao J, Cleven R, Davison W, Galceran J, Gunkel P, Kalis EJJ, Kistler D, Martin M, Noël S, Nur Y, Odzak N, Puy J, van Riemsdijk WH, Temminghoff EJM, Tercier-Waeber M-L, Toepperwien S, Town RM, Unsworth E, Warnken KW, Weng L, Xue H, Zhang H (2006) Comparison of analytical techniques for dynamic trace metal speciation in natural freshwaters. Environ Sci Technol 40:1934–1941. https://doi.org/10.1021/es051245k
Temminghoff EJM, Plette ACC, Van Eck R, Van Riemsdijk WH (2000) Determination of the chemical speciation of trace metals in aqueous systems by the Wageningen Donnan Membrane Technique. Anal Chim Acta 417:149–157. https://doi.org/10.1016/S0003-2670(00)00935-1
Ugo P, Ballarin B, Daniele S, Mazzocchin GA (1990) Electrochemistry of Yb3+ and Eu3+ at Nafion modified electrodes. J Electroanal Chem 291:187–199. https://doi.org/10.1016/0022-0728(90)87186-N
van der Stelt B, Temminghoff EJM, van Riemsdijk WH (2005) Measurement of ion speciation in animal slurries using the Donnan membrane technique. Anal Chim Acta 552:135–140. https://doi.org/10.1016/j.aca.2005.07.039
van Zomeren A, Comans RNJ (2007) Measurement of humic and fulvic acid concentrations and dissolution properties by a rapid batch procedure. Environ Sci Technol 41:6755–6761. https://doi.org/10.1021/es0709223
Weng L, Alonso Vega F, Van Riemsdijk WH (2011) Strategies in the application of the Donnan membrane technique. Environ Chem 8:466. https://doi.org/10.1071/EN11021
Weng L, Temminghoff EJM, Van Riemsdijk WH (2002) Aluminum speciation in natural waters: measurement using Donnan membrane technique and modeling using NICA-Donnan. Water Res 36:4215–4226. https://doi.org/10.1016/S0043-1354(02)00166-5
Weng L, Temminghoff EJM, van Riemsdijk WH (2001) Determination of the free ion concentration of trace metals in soil solution using a soil column Donnan membrane technique. Eur J Soil Sci 52:629–637. https://doi.org/10.1046/j.1365-2389.2001.00416.x
Yantasee W, Fryxell GE, Lin Y (2006) Voltammetric analysis of europium at screen-printed electrodes modified with salicylamide self-assembled on mesoporous silica. Anal 131:1342–1346. https://doi.org/10.1039/b609211j
Zelano IO, Cloquet C, Fraysse F, Dong S, Janot N, Echevarria G, Montargès-Pelletier E (2018) The influence of organic complexation on Ni isotopic fractionation and Ni recycling in the upper soil layers. Chem Geol 483:47–55. https://doi.org/10.1016/j.chemgeo.2018.02.023
Zelic M (2003) Electrochemical reduction of europium (3+) at increasing concentrations of different salts. Part I. Voltammetric Measurements. Croat Chem Acta 76:241–248
Acknowledgements
The authors thank Renaud Gley for his assistance during laboratory work, and Elise Rotureau for insightful discussion during experiments. We are grateful for the valuable help of Alain Mollier and Christophe Nguyen in analyzing data using R.
Funding
This work was supported by the French National Research Agency through the national program ‘Investissements d’avenir’, reference ANR-10- LABX-21–01/LABEX RESSOURCES 21. The authors also acknowledge the financial support of the region Lorraine who contributed to the post-doctoral contract of N.J.
Author information
Authors and Affiliations
Contributions
NJ: conceptualization, investigation, writing; AOF: investigation; JEG: methodology; JPP: conceptualization, methodology, supervision. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Janot, N., Groenenberg, J.E., Otero-Fariña, A. et al. Free Eu(III) Determination by Donnan Membrane Technique with Electrochemical Detection: Implementation and Evaluation. Aquat Geochem 27, 127–140 (2021). https://doi.org/10.1007/s10498-021-09392-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10498-021-09392-4