
Abstract
Transgenic plants that express double-stranded RNA (dsRNA) targeting vital insect genes have recently emerged as a valuable new tool for pest control. In this study, tobacco plants were transformed to produce dsRNA targeting Sl 102 gene that is involved in the immune response of Spodoptera littoralis larvae, a serious lepidopteran pest of several crops. Experimental larvae reared on transgenic tobacco lines showed (1) a strongly reduced level of Sl 102 transcripts, which was positively associated with food consumption; (2) a substantial impairment of the encapsulation response mediated by hemocytes; and (3) a marked increase in the susceptibility to Xentari™, a Bacillus thuringiensis-based insecticide. Importantly, this approach may allow a reduction in the doses of B. thuringiensis used for field applications and enhance its killing activity on mature larvae. The results obtained thus support the use of immunosuppressive RNAi plants to enhance the performance of microbial insecticides on lepidopteran larvae.
Similar content being viewed by others
Key message
-
RNAi-plants proved to be very effective vectors for silencing dsRNA molecules in insects.
-
Tobacco plants expressing a dsRNA targeting Sl 102 immune gene induce its silencing upon ingestion in Spodoptera littoralis larvae, a serious crop pest.
-
The resulting immune suppression enhances the killing activity of a Bt-based biopesticide.
-
Immunosuppressive RNAi-plants are environmental-friendly tools that can be used to enhance the effectiveness of Bt sprays or other entomopathogen-based insecticides in integrated pest management strategies.
Introduction
Currently, recombinant DNA technology is routinely used for the production of genetically modified crops (also known as biotech crops) to enhance plant defenses against abiotic and biotic stress (Ricroch and Hénard-Damave 2016). Since their introduction in 1996, the area under biotech crop cultivation has increased more than 100-fold, reaching 191.7 million hectares in 2018 distributed across 26 countries worldwide, while another 44 countries imported these biotech crops (http://www.isaaa.org). Although the market for insect-resistant crops is dominated by plants expressing toxins from the entomopathogen Bacillus thuringiensis (i.e., Cry and Vip toxins alone or combined by gene pyramiding) (Anderson et al. 2019), it is worth mentioning that several efforts to introduce other effective transgenes into plants have been made.
Several “antinutritional” proteins (i.e., that impair digestion and/or absorption of nutrients), for instance, have been considered excellent options for expression in plant tissues to confer protection against phytophagous insects. Genes encoding for protease inhibitors (PIs) have been claimed to be good candidates for crop engineering (Dunaevsky et al. 2005; Schlüter et al. 2010). PIs are key defense metabolites produced by plants to fight against herbivorous insects, and their mode of action involves the impairment of protein digestion, which in turn causes retarded growth or even death (Macedo et al. 2015). The expression of exogenous PIs in plants, in particular serine- and cysteine-PIs, has been proven to confer resistance to Lepidoptera and Coleoptera, respectively (Dunaevsky et al. 2005; Schlüter et al. 2010). Among plant defense metabolites with antinutritional properties, lectins have proven to be very effective especially against Hemiptera (Macedo et al. 2015). The expression of lectins in plants inhibits larval growth and induces moderate mortality in Lepidoptera while causing high mortality in hemipteran insects by different mechanisms (Vasconcelos and Oliveira 2004; Shahidi-Noghabi et al. 2010a, b; Vandenborre et al. 2011; Caccia et al. 2012; Macedo et al. 2015). Interest in lectins was also motivated by their ability to act as vectors for the delivery of insecticidal molecules with hemolymphatic targets, such as GNA-venom fusion proteins (Rao et al. 1998; Foissac et al. 2000; Sétamou et al. 2002; Wang et al. 2005; Nagadhara et al. 2004; Trung et al. 2006; Fitches et al. 2012; Mi et al. 2017), an approach that has also been exploited in plants for the expression of chimeric proteins (Liu et al. 2016; Nakasu et al. 2014; Rauf et al. 2019). It is worth mentioning the exploitation of the antinutritional properties of antivitamins (i.e., compounds that interfere with the biological function of vitamins) to develop insect-resistant plants. Avidin, a glycoprotein present in avian egg white, forms complexes with biotin (vitamin B12), and this binding impedes molecular recognition and uptake of the vitamin, inducing strong biotin deficiency. The expression of avidin in transgenic maize, tobacco and wheat indeed conferred resistance against many species of stored-product coleopteran and phytophagous lepidopteran pests (Kramer et al. 2000; Burgess et al. 2002; Abouseadaa et al. 2015). In addition, some chitinolytic enzymes proved to be interesting candidates for in planta crop protection, as they do not affect non-target organisms that lack chitin (i.e., vertebrates and plants), but are active against many noxious insect pests (Wang et al. 1996; Ding et al. 1998; Corrado et al. 2008; Berini et al. 2018). By altering peritrophic matrix architecture, these enzymes affect the efficiency of macronutrient digestion in the insect midgut (Berini et al. 2018).
Undoubtedly, the so-called RNAi-plants represent the most promising new generation of insect-resistant plants. Discovered in 1998 in a nematode (Fire et al. 1998), RNA interference (RNAi) (i.e., the mechanism of gene silencing mediated by dsRNA molecules) has been proven to be almost ubiquitous in eukaryotes wherein it represents a mechanism of gene regulation and a key player in antiviral defense (Agrawal et al. 2003; Kim and Rossi 2008; Ghildiyal and Zamore 2009; Schuster et al. 2019). RNAi has been exploited as a valuable tool to study gene function in many organisms and for many other purposes (Agrawal et al. 2003; Zhu and Palli 2020), such as the production of insect-resistant plants expressing dsRNA molecules (Mamta and Rajam 2017; Zhang et al. 2017; Zhu and Palli 2020). When plant tissues expressing dsRNA targeting essential insect genes (e.g., genes encoding V-type ATPase subunits, acetylcholinesterase, actin and tubulin isoforms, and enzymes involved in detoxification processes or endocrine regulation) are ingested, the silencing induces growth retardation or even death of the insect (Baum et al. 2007; Mao et al. 2011; Zhang et al. 2015; Eakteiman et al. 2018; Guo et al. 2018; Fu et al. 2020). A proof-of-concept study was published in 2007 by Baum and colleagues that demonstrated the insecticidal effect of transgenic corn expressing dsRNA against the V-ATPase gene of Diabrotica virgifera virgifera, resulting in a decrease in feeding damage and an increase in crop protection (Baum et al. 2007). The great interest in RNAi-crop technology for insect control resides in the very targeted delivery (dsRNA molecules can be expressed in specific plant tissues that are damaged by the insect pest) and specificity of interfering dsRNA to phytophagous insects (that can reach the species and, in some cases, even the strain level or even a specific developmental stage); this specificity results in no off-target effects and increased environmental safety (Fletcher et al. 2020).
In addition to the direct killing of the pest, the versatility of RNAi plants has been demonstrated by Mao and colleagues (Mao et al. 2007, 2011). Indeed, in the cotton bollworm Helicoverpa armigera, ingestion of dsRNA targeting a cytochrome P450 monooxygenase gene (i.e., CYP6AE14) increases the toxic effects of the cotton metabolite gossypol which is normally tolerated by this pest due to detoxification mediated by different enzymes (Mao et al. 2007). Cotton plants expressing dsRNA targeting CYP6AE14 drastically impaired insect growth and significantly reduced plant damage (Mao et al. 2011). In the present work, we exploited plants as vectors for immunosuppressive dsRNA molecules to enhance insect pest susceptibility to entomopathogens. In particular, tobacco plants were transformed to express interfering dsRNA that specifically targeted Sl 102 Spodoptera littoralis gene (a gene involved in immune cellular responses, as microbial nodulation, in S. littoralis larvae) (Di Lelio et al. 2014; Caccia et al. 2016) and, were able to increase insect pest susceptibility against the entomopathogen B. thuringiensis. The results obtained in the present work have thus important implications for the effectiveness of microbial control agents (MCAs) in integrated pest management (IPM) strategies.
Materials and methods
Molecular cloning
Total RNA extracted from S. littoralis hemocytes was subjected to retrotranscription (Ambion® RETROscript® Kit, Thermo Fisher Scientific, Waltham, Massachusetts, USA) and, then, used for PCR amplification of a 479 bp Sl 102 fragment, with specific primers (i.e., Sl102-attB1 Fw and Sl102-attB2 Rv in Table 1 of the Supplementary material).
Recombinant plasmid molecules were obtained by Gateway® Recombination Cloning Technology (Thermo Fisher Scientific). An entry clone was generated by performing a BP recombination reaction between the attP-containing pDONR™/Zeo vector (Thermo Fisher Scientific) and the attB PCR product. The reaction mixture included 20–40 fmol of PCR products with attB sites, 150 ng of Gateway™ pDONR™/Zeo Vector (Thermo Fisher Scientific), 2 μl of BP Clonase® enzyme (Thermo Fisher Scientific) and TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0) to a final volume of 8 μl. The mixture was incubated at room temperature overnight. Subsequently, 1 μl of Proteinase K (Thermo Fisher Scientific) was added, and samples were incubated at 37 °C for 10 min. For the creation of Gateway® Expression Clones, LR reactions were performed using 250 ng of attL-flanked entry clone, 150 ng of destination vector (containing attR sites), 2 μl of LR Clonase® enzyme and TE buffer to a final volume of 8 μl. The reaction mixture was incubated at room temperature overnight. The destination vector was pHellsgate12 (CSIRO Plant Industry, Canberra, Australia), a Gateway® adapted binary vector suitable for gene silencing in plants (Helliwell et al. 2002). Plasmid DNA was isolated using the alkaline lysis protocol of the Purelink™ Quick Plasmid Miniprep Kit (Thermo Fisher Scientific), quantified by densitometry with agarose gel electrophoresis (0.8% w/v) and screened by PCR for the presence and orientation, if necessary, of the inserts (Sambrook and Russell 2001). The sequence of the inserted DNA was verified by Sanger sequencing, performed at the Bio-Fab Research Company (Rome, Italy). The recombinant plasmids were introduced into Escherichia coli cells by electroporation at 2.5 kW for 5 milliseconds or heat shock (Sambrook and Russell 2001). Following transformation, the suspension of E. coli cells was placed on ice and 250 μl of Super Optimal broth with Catabolite repression (SOC) medium (Thermo Fisher Scientific) (Sambrook and Russell 2001) were added. Cells were then incubated at 37 °C for 30 min with agitation (150 rpm) in Luria–Bertani (LB) broth and plated on LB-agar with the appropriate antibiotic(s).
DNA samples were amplified in a final volume of 20 μl containing 0.5 U of Taq DNA polymerase (Promega, Milan, Italy), 1 × buffer (Promega), 1.5 mM MgCl2, 10 mM dNTPs and 20 pmol of each primer (forward and reverse). The amount of DNA used as the template ranged from 10 ng (for the amplification of plasmid DNA templates) to 100 ng (for the amplification of plant genomic DNA). Colony PCR was also used in the screening for recombinant plasmids (Sambrook and Russell 2001). PCR products were verified by agarose gel (1% w/v) electrophoresis (Sambrook and Russell 2001). The marker used was a 1 kb Plus DNA ladder (Thermo Fisher Scientific). Primers were obtained from Thermo Fisher Scientific, and sequences are reported in Table 1 of the Supplementary material. Reactions were carried out in a Gene Amp 2700 PCR cycler (Thermo Fisher Scientific).
All primer pairs were designed using Primer Express 3.0 software (Thermo Fisher Scientific) following the standard procedure.
Plant transformation
The binary vector carrying Sl 102 dsRNA was produced using Gateway® technology and mobilized in Agrobacterium tumefaciens. Tobacco (Nicotiana tabacum L., ‘Samsun’ NN) transformation and regeneration of kanamycin-resistant plants were carried out as previously described (Corrado et al. 2016). Putative transgenic plants were transferred in sterile soil and grown under controlled conditions (T0 generation). Transgenic plants of the T1 generation were identified by successive rounds of selection in kanamycin containing media and PCR analyses.
Molecular characterization of transgenic plants
DNA from putative transgenic plants was extracted according to Fulton’s protocol (Fulton et al. 1995). Transgene presence was verified by PCR targeting the Sl 102 gene (for primer sequences see Table 1 of the Supplementary material). Total RNA was isolated from the leaves of 4 week-old plants, first strand cDNA was synthesized, and qRT-PCR was performed as previously reported (Corrado et al. 2012) to detect the production of Sl 102 dsRNA. Relative quantification was carried out using the ΔΔCt method (Livak and Schmittgen 2001; Pfaffl 2001; Pfaffl et al. 2002) using two technical replicates for each of the 3 biological replicates per sample. The housekeeping gene elongation factor 1 α (EF-1α) was used as an endogenous reference gene for the normalization of the expression levels of the target transcript. The expression analysis was carried out on the T0 generation and later on the T1 generation using a genotype with a low level of transgene expression as calibrator. Sequences of the primers used for qRT-PCR (i.e., Sl102 Nt Rt Fw, Sl102 Nt Rt Rv, EF-1α Rt Fw, and EF-1α Rt Rv) were designed using Primer Express 3.0 software (Thermo Fisher Scientific), following the standard procedure, and are reported in Table 1 of the Supplementary material.
Insect rearing and preparation of experimental larvae for feeding bioassays
S. littoralis larvae were reared on an artificial diet (41.4 g/l wheat germ, 59.2 g/l brewer’s yeast, 165 g/l corn meal, 5.9 g/l ascorbic acid, 1.53 g/l benzoic acid, 1.8 g/l methyl 4-hydroxybenzoate and 29.6 g/l agar), at 25 ± 1 °C, 70 ± 5% relative humidity, and under a 16:8 h light–dark period.
All feeding bioassays on plant tissues described below were carried out in triplicate under the same environmental conditions reported above and using S. littoralis larvae that were maintained until the beginning of the experiment on subapical leaves of 4 week-old wild-type (WT) tobacco plants. These leaves were replaced daily in plastic boxes (30 × 40 × 15) bottom lined with 50 ml of 1.5% (w/v) agar in water to preserve leaf turgor. The experimental larvae were checked daily and those molting within 4 h formed experimental groups of synchronized larvae.
Time course of Sl 102 gene silencing and immune suppression
Different time course experiments were carried out on S. littoralis larvae fed transgenic plant leaves expressing Sl 102 dsRNA to assess the level of silencing of the targeted gene. All the described experiments were carried out in plastic boxes, prepared as described above.
In the first time course experiment, S. littoralis eggs were left to hatch on WT tobacco leaves and larvae fed on those leaves for 12 h. First instar larvae were then grouped (100 larvae per box) and fed transgenic plant lines or WT tobacco (controls) leaves. Larvae were synchronized daily as described above, and at the end of the third instar (i.e., just before molting), experimental larvae were divided into additional boxes (25 larvae per box) to prevent cannibalism, and reared until pupation on experimental tobacco leaves. For total RNA extraction, whole larvae (fully grown second, third, fourth, and fifth instars, those on the first day of the sixth instar and those in the prepupal stage, i.e., 24 h after feeding cessation) were collected into TRIzol® reagent (Thermo Fisher Scientific). In the second time course experiment, S. littoralis eggs were left to hatch on WT tobacco leaves and larvae fed on WT tobacco leaves until the end of the third instar. Then, 150 newly molted fourth instar S. littoralis larvae (25 larvae per box) were reared on transgenic tobacco leaves (or on WT in control experiments) and allowed to develop until the pupal stage. The experimental larvae, i.e., those fully grown to the fourth and fifth instars, on the first day of the sixth instar and in the prepupal stage were processed to collect hemocytes into TRIzol® reagent and perform total RNA extraction (Di Lelio et al. 2019).
Silencing efficiency was evaluated by qRT-PCR, as described below, and the impact of gene silencing on cellular immune competence was assessed by measuring the encapsulation index of chromatography beads in fifth instar larvae fed, from the fourth instar, with the experimental tobacco leaves as described above. The encapsulation assay was carried out as previously described (Di Lelio et al. 2014; Becchimanzi et al. 2019).
RNA extraction and qRT-PCR
Total RNA was extracted from the whole larval body or from hemocytes in TRIzol® reagent (Thermo Fisher Scientific), according to the manufacturer’s instructions. The concentration and purity of total RNA were determined using a Varioskan™ Flash Multimode Reader (Thermo Fisher Scientific). Transcription of the Sl 102 gene (Accession Number KJ544881.1) was measured by one-step qRT-PCR, using the SYBR Green PCR Kit (Thermo Fisher Scientific), according to the manufacturer’s instructions.
Relative qRT-PCR was carried out using Sl 102 gene-specific primers (see Table 1 in Supplementary material for Sl102 Rt Fw and Sl102 Rt Rv sequences), which were designed to detect a segment of the Sl 102 mRNA outside to the segment targeted by the dsRNA. The S. littoralis β-actin gene (Accession Number Z46873) was used as an endogenous control for RNA loading (primer sequences are reported in Table 1 of the Supplementary material). All primer pairs were designed using Primer Express 3.0 software (Thermo Fisher Scientific), following the standard procedure. Negative controls (water) were included in each run of the qRT-PCR. The amount of target transcript relative to the endogenous control was determined using the ΔΔCt method (Livak and Schmittgen 2001; Pfaffl 2001; Pfaffl et al. 2002). For validation of the ΔΔCt method the difference between the Ct value of Sl 102 and the Ct value of β-actin transcripts [ΔCt = Ct (Sl 102) − Ct (β-actin)] was plotted versus the log of tenfold serial dilutions (5000, 500, 50, 5 and 0.5 ng) of the purified RNA samples. The plot of log total RNA input versus ΔCt displayed a slope of less than 0.1 (Slope = 0.0154, R2 = 0.0776), indicating that the efficiencies of the two amplicons were approximately equal.
Sl 102 silencing by transgenic tobacco and effects on Bt killing activity
A preliminary experiment was performed to determine the minimum feeding time on transgenic tobacco leaves to obtain gene silencing in the larvae used in bioassays, i.e., fourth and fifth instar larvae. Larvae were reared on WT tobacco leaves as described previously. Sixty-four newly molted fourth and fifth instar larvae were then transferred to transgenic tobacco leaves (4 and 6.25 cm2 leaf squares were offered to fourth and fifth instar larvae, respectively) and silencing was monitored over time by qRT-PCR as previously described, using total RNA extracted from hemocytes collected in TRIzol® reagent (Di Lelio et al. 2019). Control larvae were fed on WT tobacco leaves. To search for a correlation between the silencing level of every single larva, tobacco leaf pieces were collected after 14 h and the respective leaf consumption was arbitrarily defined as high, medium, or low.
For the bioassays with Bt bioinsecticide, synchronous fourth and fifth instar larvae, reared on WT tobacco plants or on the two transgenic lines, were obtained as described above, and singly transferred to multiwell plastic rearing trays (RT32W, Frontier Agricultural Sciences, USA) that were bottom-lined with 1 ml of 1.5% (w/v) agar in water, to keep the leaf squares turgid. The rearing wells, each containing a leaf square and a larva, were closed by perforated plastic lids (RTCV4, Frontier Agricultural Sciences, USA). The leaf squares were obtained by cutting leaves uniformly sprayed with Xentari™ (Valent BioSciences), a bioinsecticide based on Bt subsp. aizawai, that contains several Cry toxins and active on Spodoptera spp. Experimental tobacco leaves sprayed with distilled water were used as controls.
Preliminary bioassays were performed to determine the sublethal doses (i.e., causing growth retardation and/or weight decrease but no mortality) of Xentari™ in controls (larvae fed on WT tobacco). Sublethal doses were 1 and 3 μg/cm2, for fourth and fifth instar larvae, respectively.
In the first bioassay (sequential treatment), for each experimental tobacco line, newly molted fourth instar larvae were fed ad libitum on leaves for 3 days. Then, 64 larvae were transferred to multiwell plastic rearing trays prepared as described above and fed ad libitum for 3 days with leaves of each tobacco plant line treated with Xentari™ or water as controls. Mortality was recorded daily from the beginning of the bioassay for 8 days, and the weight of the surviving larvae was recorded on Day 8.
Two other bioassays (concurrent treatment with Bt) were carried out on fourth and fifth instar larvae. Both concurrent bioassays were carried out by feeding fourth instar or fifth instar larvae leaf pieces from each experimental tobacco plant line for 24 h and then treating them with Bt for the following 3 days. Mortality was recorded daily from the beginning of the bioassay for 6 days, and the weight of the surviving larvae was recorded on Day 6.
Statistical analysis
Sl 102 dsRNA expression in tobacco transgenic lines was analyzed using unpaired Student’s t test. Sl 102 gene expression in larvae and data from the encapsulation assay were analyzed using one-way ANOVA, followed by Tukey’s multiple-comparison post hoc test. Normality of the data was checked by using the Shapiro–Wilk test and the D’Agostino-Pearson test, while homoscedasticity was checked with Bartlett’s test. When ANOVA assumptions were not fulfilled (as for larval weight), nonparametric Kruskal–Wallis ANOVA followed by Dunn’s multiple comparisons post-hoc test was used. Survival curves of S. littoralis larvae were compared using Kaplan–Meier and log-rank analysis. All data were analyzed using GraphPad Prism, version 6.0b (GraphPad software; San Diego, California, USA).
Results
Production of tobacco plants expressing Sl 102 dsRNA
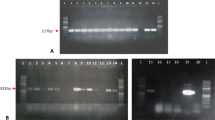
For the creation of an expression vector containing the construct to produce a dsRNA hairpin using the Gateway® Cloning System, a sequence of the Sl 102 gene was amplified with Sl102-attB1 Fw and Sl102-attB2 Rv primers using RNA extracted from S. littoralis hemocytes as the template. Purified PCR products were then recombined with the pDONOR™/Zeo vector to obtain the pDSl102 entry clone. pDSl102 was then recombined with the destination vector pHellsgate12, yielding the pHSl102 vector with the Sl 102 dsRNA hairpin expression cassette (Fig. 1a). Recombinant pHSl 102 vectors were checked by PCR to ensure the presence of Sl 102 fragments in both orientations (Fig. 1a and b) and used for stable tobacco transformation. Regenerated plants were transferred in vivo to an isolated growing chamber. The DNA of putatively transformed plants was isolated and analyzed by PCR using the AGRI64(2) Fw and attB2 Rv (Fig. 1a in Supplementary material), and the AGRI51(2) Fw and attB1 Rv (Fig. 1b in Supplementary material) primer combinations to check the presence and correct orientation of the Sl 102 sequences. As expected, the expression analysis of the transgenic plants produced (T0 generation) revealed genotypes showing different levels of transgene expression (Fig. 2 in Supplementary material). According to the transgene expression levels and to test a more stable genetic background, two transgenic genotypes were selected to obtain T1 generations for subsequent investigations. Figure 2 shows the relative quantification of transgene expression on experimental transgenic tobacco plants (Line 1 and Line 2) and their phenotype compared to WT tobacco (Fig. 2a and b, respectively).

Production of the Sl 102 hairpin using the Gateway® cloning system. a Schematic representation of the expression cassette of the pHSl102 construct used for tobacco transformation (not to scale). P35S: 35S RNA CaMV promoter sequence; Sl 102 insert: Sl 102 sequence flanked by two attR recombination sites in the sense orientation; PdK: pyruvate orthophosphate dikinase intron in the sense orientation fused to the castor bean catalase-1 intron in the antisense orientation; Sl 102 insert: Sl 102 sequence flanked by two attR recombination sites in the antisense orientation; Pnos: nopaline synthase gene promoter; nptII: neomycin phosphotransferase II coding sequence; Tnos: nopaline synthase gene terminator. In a the AGRI primers to detect the sense (AGRI 51–56) and the antisense (AGRI 64–69) Sl 102 fragments by PCR screening are shown. b Example of DNA fragment analysis using agarose gel electrophoresis of recombinant plasmids. Lanes 1 and 4: no template control; Lanes 2 and 3: PCR products of putative recombinant pHSl102 with the AGRI 51–56 primers; M: DNA ladder; Lanes 5 and 6: PCR products of putative recombinant pHSl102 with the AGRI 64–69 primers. Primers sequences are reported in Table 1 of the Supplementary material

Transcriptional analysis of Sl 102 dsRNA by qRT-PCR in transgenic lines (a). The two transgenic plant lines showing the highest expression of Sl 102 dsRNA (Line 1 and Line 2) were used in feeding bioassays. Transcript quantities were calibrated based on the transgenic genotype showing the lowest expression, which was arbitrarily set as 1 (*P = 0.000265; **P = 0.0013, Student’s t test). The transgenic lines used in the experiments did not show phenotypic alterations with respect to wild type (WT) tobacco (b)
Silencing and immunosuppressive effects of Sl 102 dsRNA transgenic plants
Both transgenic tobacco lines expressing Sl 102 dsRNA induced a significant level of gene silencing when fed to S. littoralis larvae (Fig. 3a and b).

Gene silencing in Spodoptera littoralis larvae reared on wild-type (WT) or Sl 102 dsRNA-expressing (Line 1 and Line 2) tobacco plant leaves. Larvae were maintained on WT tobacco leaves and fed on transgenic plant leaves from the first (a) or fourth instar (b), as shown in the experimental plan above each histogram. Control larvae were reared on WT tobacco leaves. The comparison of the mean values was performed within each developmental instar, and those that were significantly different are denoted with different letters (*P < 0.0001, one-way ANOVA)
When the larvae were fed precociously with leaves of transgenic tobacco plants (from the first instar), a significant level of gene silencing was observed only when the experimental larvae attained the fourth instar (one-way ANOVA: second instar—F(2, 25) = 2.60; P = 0.00943, dF = 27; third instar—F(2, 26) = 0.483; P = 0.6223; dF = 28; fourth instar—F(2, 23) = 659.3, P < 0.0001, dF = 25) (Fig. 3a). This reduction in the Sl 102 gene transcription rate was consistently observed throughout the remainder of the bioassay, up to the prepupal stage (one-way ANOVA: fifth instar—F(2, 26) = 488.5, P < 0.0001, dF = 28; sixth instar—F(2, 23) = 1114.0, P < 0.0001, dF = 25; prepupal stage—F(2, 24) = 8.0, P < 0.0001, dF = 26) (Fig. 3a).
To check the hypothesis that the onset of gene silencing was influenced by the low amount of plant tissue (and thus of silencing Sl 102 dsRNA) ingested by the earliest instar larvae, we performed the feeding bioassay starting with fourth instar larvae (Fig. 3b). The results supported our hypothesis, indeed, the occurrence of gene silencing was already observed at the end of the fourth instar and was statistically significant for all the following time points considered (one-way ANOVA: fourth instar—F(2, 82) = 124.8; P < 0.0001; dF = 84; fifth instar—F(2, 81) = 240.6; P < 0.0001; dF = 83; sixth instar—F(2, 56) = 665.6; P < 0.0001; dF = 58; prepupal stage—F(2, 44) = 187.6; P < 0.0001; dF = 46) (Fig. 3b). Moreover, gene silencing was measured in detail during the 3 days of fourth instar, demonstrating that although larvae do not feed continuously during the intermolt period, the significance of the silencing effect was maintained (14 h—Kruskal–Wallis: KW = 10.24; P = 0.006; 24 h—one-way ANOVA: F(2, 52) = 229.7; P < 0.0001; dF = 54; 38 h—Kruskal–Wallis: KW = 42.94; P < 0.0001; 48 h—Kruskal–Wallis: KW = 42.34; P < 0.0001; one-way ANOVA: 62 h—F(2, 56) = 1733; P < 0.0001; dF = 58; one-way ANOVA: 72 h—F(2, 57) = 645.5; P < 0.0001; dF = 58) (Fig. 3 in the Supplementary material).
The level of Sl 102 silencing observed was expected to induce an immunosuppressed phenotype, characterized by the impairment of both nodulation and encapsulation responses by hemocytes (Di Lelio et al. 2014; Caccia et al. 2016, 2020). Indeed, encapsulation and melanization reactions in silenced fifth instar larvae collected from the previous experiment (Fig. 3b) were significantly affected compared with those observed in control larvae fed WT tobacco (one-way ANOVA: F(2, 112) = 4568; P < 0.0001; dF = 114) (Fig. 4). In particular, 24 h after hemocoelic injection, chromatographic beads recovered from the hemocoel of control larvae were completely encapsulated and melanized by hemocytes (Encapsulation Index, EI = 87.0%), while this cellular immune response completely failed in larvae fed the Line 1 (EI = 16.7%,) and Line 2 (EI = 18.0%) transgenic plant leaves.

Encapsulation index of Spodoptera littoralis larvae reared on wild type (WT) (controls) or Sl 102 dsRNA-expressing (Line 1 and Line 2) tobacco plant leaves. The encapsulation response was significantly inhibited in S. littoralis larvae showing Sl 102 silencing. Chromatography beads collected from the hemolymph of control larvae were completely encapsulated and melanized while hemocyte capsule formation was not observed in larvae fed transgenic plant lines. Mean values with different letters are significantly different (P < 0.0001, one-way ANOVA)
Influence of leaf consumption on the silencing effect of Sl 102 dsRNA transgenic plants
Prior to performing bioassays with Bt bioinsecticide, the minimum time interval of feeding required to induce a significant level of gene silencing in larval stages used for the bioassays (fourth and fifth instars) was assessed. Larvae were thus allowed to feed on plant tissue for 14 and 24 h. For both instars silencing was significant only after 24 h (one-way ANOVA: fourth instar—F(2, 93) = 279.6, P < 0.0001; dF = 95; fifth instar—F(2, 93) = 525.7; P < 0.0001; dF = 95) (Fig. 5a and b for fourth and fifth instars, respectively). Indeed after 14 h the silencing was not significant due to the very high variability in the rates of Sl 102 transcription among larvae (Kruskal–Wallis test: fourth instar—KW = 5.769; P = 0.056; fifth instar—KW = 2.258; P = 0.3234) (Fig. 5a and b for fourth and fifth instar, respectively).

Relative expression of the Sl 102 gene in fourth and fifth instar larvae. The Sl 102 transcript level was not affected after 14 h in larvae fed transgenic tobacco lines for either fourth instar larvae (P = 0.056, Kruskal–Wallis test) (a) or fifth instar larvae (P = 0.3234, Kruskal–Wallis test) (b). A significant reduction in Sl 102 gene expression resulted after 24 h of feeding on transgenic plants for fourth instar larvae (a) and fifth instar larvae (b) (P < 0.0001, one-way ANOVA)
To check whether this variability was related to the amount of leaf consumption, the transcription data related to the 14 h time-point were grouped on the basis of leaf consumption. A clear dose–response effect on gene silencing was observed for both fourth instar larvae (one-way ANOVA: low leaf consumption level—F(2, 60) = 2.67; P = 0.0778; dF = 62; medium leaf consumption level—F(2, 47) = 51.11; P < 0.0001; dF = 49, high leaf consumption level—F(2, 43) = 170.0; P < 0.0001; dF = 45) (Fig. 6a) and fifth instar larvae (one-way ANOVA: low leaf consumption level—F(2, 55) = 0.29; P = 0.7488; dF = 57; medium leaf consumption level—Kruskal–Wallis test: KW = 35.32; P < 0.0001; high leaf consumption level—one-way ANOVA: F(2, 41) = 165.8; P < 0.0001; dF = 43) (Fig. 6b).

Relative expression of the Sl 102 gene in fourth and fifth instars larvae after 14 h, as affected by leaf consumption. The expression of the Sl 102 gene was significantly lower, both for fourth (a) and fifth (b) instar larvae, only for medium and high leaf tissue consumption, compared to controls. Mean values with an asterisk are significantly different (*P < 0.0001, one-way ANOVA or Kruskal–Wallis test)
Sl 102 dsRNA-expressing plants enhance the killing activity of Bacillus thuringiensis
The high effectiveness of transgenic plants in the induction of Sl 102 silencing and immune suppression prompted us to assess whether silenced S. littoralis larvae were more susceptible to a treatment with a Bt-based biopesticide (Xentari™), as previously reported in the case of Sl 102 dsRNA delivered with different methods (Caccia et al. 2016, 2020) and of Sl gasmin, another immune gene (Di Lelio et al. 2019).
To take into account the potential discrepancy in Xentari™ efficacy when the bioinsecticide is used against larvae that have already fed and developed on a transgenic crop or larvae that come into contact with transgenic tobacco and the bioinsecticide more or less simultaneously, different bioassays were performed. Moreover, since the aim of the bioassays was measuring the effect of RNAi plants on Xentari™ efficacy, doses that were sublethal to controls, i.e., larvae fed WT plants, were used (see “Sl 102 silencing by transgenic tobacco and effects on Bt killing activity” in Material and Methods for experimental details).
In the first set of experiments (sequential treatments), fourth instar S. littoralis larvae were alimented for 3 days on leaf squares of transgenic plants expressing Sl 102 dsRNA and on Day 4 they were exposed with Xentari™ for 3 subsequent days, and maintained on transgenic leaf tissues until Day 8. Xentari™ induced significantly higher levels of mortality in larvae fed with Sl 102 dsRNA-transgenic plants compared to controls (log-rank test: χ2 = 271.5, P < 0.0001, dF = 5) (Fig. 7a). Moreover, surviving larvae showed a significant reduction in the weight increase (one-way ANOVA: F(5, 199) = 448.7, P < 0.0001; dF = 204) (Fig. 7d).

Bioassay with Spodoptera littoralis larvae. In a fourth instar larvae were fed Sl 102 dsRNA-transgenic plants before Bt treatment. Newly molted fourth instar larvae were alimented for 3 days with leaf squares from the experimental tobacco plants and then, soon after molting to the fifth instar, with 3 μg/cm2 of Xentari™ for 3 additional days (see “Materials and methods” section for experimental details). In b, c larvae were concurrently exposed to dsRNA and Bt. Newly molted fourth instar (b) or fifth instar (c) larvae were alimented for 24 h with untreated experimental plant leaves and, for the following 3 days, with leaf squares sprayed with 1 μg/cm2 (b) or 3 μg/cm2 (c) of Xentari™, which were sublethal doses for larvae fed WT tobacco (see the “Materials and methods” section for experimental details). Survival was monitored until Day 8 (a) or Day 6 (b, c) when the weight of the surviving experimental larvae was assessed (d–f). The timing of Bt treatments is indicated by arrows. The values reported are the mean ± standard errors: curves that significantly differ from controls are indicated with an asterisk in (a–c) (P < 0.0001, log-rank Mantel–Cox test) and with different letters in (d–f) (P < 0.0001, Kruskal–Wallis test)
Based on the results obtained above (Fig. 5 and Fig. 3 in Supplementary material), after 24 h of feeding on transgenic plants, a significant level of gene silencing in fourth and fifth instar larvae was observed; thus, Bt treatment was performed on leaf squares for 3 consecutive days, starting 24 h after the onset of the bioassay. For both fourth and fifth instar larvae, the administration of Sl 102 dsRNA-transgenic plants and Xentari™ caused significantly higher mortality in Sl 102-silenced larvae compared to controls (log-rank test—fourth instar larvae—χ2 = 235; P < 0.0001; dF = 5; fifth instar larvae—χ2 = 230.2; P < 0.0001; dF = 5) (Fig. 7b and c). This was accompanied by a significant developmental impairment for both fourth instar (Kruskal–Wallis: KW = 119.0; P < 0.0001) (Fig. 7e) and fifth instar larvae (Kruskal–Wallis: KW = 135.1; P < 0.0001) (Fig. 7f).
Upon silencing, older instar larvae are thus more susceptible to Bt treatment, with minor changes depending on the duration of the feeding period on dsRNA-expressing plants. Indeed, although 24 h of feeding on Sl 102 dsRNA-transgenic lines was sufficient to obtain an increase in Xentari™ effectiveness in fifth instar larvae (Fig. 7c), larvae fed on transgenic plants for all fourth instar resulted in significantly higher mortality on the last day (one-way ANOVA: F(2, 6) = 8.85, P = 0.0162, dF = 8 for Line 1; F(2, 6) = 15.28, P = 0.0044, dF = 8 for Line 2) (Fig. 7a, Table 2 in the Supplementary material).
Regardless of the administration protocol, bioassays clearly show that the application of the Bt formulate causes in a few days a dramatic drop of survival (around −50%) only in larvae fed transgenic tobacco lines expressing Sl 102 dsRNA (Fig. 7a, b and c).
Discussion
Although a number of potential traits have been tested for plant engineering against insect pests, the expression of Bt toxins remains the most widely used (Anderson et al. 2019) and RNAi-plants are the focus of most research efforts and market considerations (Zhang et al. 2017; Anderson et al. 2019; Fletcher et al. 2020; Liu et al. 2020). The reasons for the lack of success of the “antinutritional” traits appear evident in some cases. For instance, although early achievements in PI-expressing plants were encouraging, coevolution between insect herbivores and host plants has led to the development of reciprocal adaptations (Zhu-Salzman and Zeng 2015; Singh et al. 2020). The ability of insects to cope with plant defense metabolites by a variety of mechanisms (e.g., remodeling of the digestive proteasome, expression of inhibitor-insensitive proteases, inactivation of PI) has thus hampered interest and discouraged further research on this strategy (Zhu-Salzman and Zeng 2015; Singh et al. 2020). On the other hand, the use of feed or food based on transgenic crops with antivitamins, such as avidin, was not favored. These compounds combine with vitamins reducing their availability; thus, supplementation of the vitamins in the diet or processing of the crop for the release of the vitamins from complexes is required. In addition, since a thorough risk assessment is still lacking, the presence of antivitamins in crops may suffer from a lack of public acceptability. The low success of lectin-expressing plants is likely because these proteins are potential food allergens (Barre et al. 2020). Although they proved to be very effective against pests, especially when fused with proteins from insect specific and even insect pest-specific arthropod venoms, no impact on beneficial insects has been observed (Gatehouse et al. 2011; Nakasu et al. 2014).
It is worth mentioning that the in planta coexpression of genes encoding insecticidal molecules with different modes of action can strongly enhance control effectiveness and delay the development of pest resistance (Sainsbury et al. 2012; Dormatey et al. 2020). This strategy (i.e., stacking or pyramiding), which is now routine for commercialized Bt-crops expressing multiple toxins, has also been attempted for antinutritional molecules (Boulter et al. 1990; Abdeen et al. 2005; Senthilkumar et al. 2010). Furthermore, plants have been transformed to express protease inhibitors or lectins with Bt toxins, leading to an enhancement of protection against both chewing and sucking insects (Fan et al. 1999; Maqbool et al. 2001; Su et al. 2011; Zhang et al. 2011; Boddupally et al. 2018).
Herein we have attempted a novel strategy of using transgenic plants as shuttles for molecules that can prepare insects for treatment with microbial control agents (MCAs) such as B. thuringiensis. The rationale behind the present work relies on the evidence that immunosuppressed insects are more susceptible to entomopathogens. Chen and coworkers have demonstrated that recombinant Isaria fumosorosea fungi expressing dsRNA targeting an immune gene (i.e., Toll-like receptor 7) in Bemisia tabaci are more virulent against this pest (Chen et al. 2015). Similarly, RNAi silencing of AgraRelish, a transcription factor of the IMD pathway in the Coleoptera Anthonomus grandis, improves the efficacy of the fungus Metarhizium robertsii (Moreira-Pinto et al. 2021). Interestingly, molecules of parasitic origin that impair immune responses in lepidopteran larvae have been proven to enhance entomopathogen performance against these insects. rVPr1, a protein from the venom of the endoparasitic wasp Pimpla hypochondriaca that affects hemocyte responses in several lepidopteran species, was able to enhance the susceptibility of Lacanobia oleracea and Mamestra brassicae larvae to B. thuringiensis and Beauveria bassiana (Dani et al. 2004; Richards et al. 2011, 2013; Richards and Dani 2010). The use of polydnaviruses or polydnavirus-derived factors to disrupt immune responses resulted in an increase of baculovirus infection in S. littoralis and Manduca sexta larvae (Rivkin et al. 2006; Washburn et al. 2000). Moreover, previous studies from our group have demonstrated that immunosuppressed S. littoralis larvae are more susceptible to B. thuringiensis. In particular, silencing of the Sl 102 and Sl gasmin immune genes, both individuated in the framework of studies on host-parasitoid interactions (Di Lelio et al. 2014, 2019; Gasmi et al. 2015), led to an increase in the mortality of larvae treated with B. thuringiensis or Bt toxins (Caccia et al. 2016, 2020; Di Lelio et al. 2019).
This evidence prompted us to use transgenic plants as vectors for immune suppressive molecules to impair the response of a phytophagous pest against entomopathogens. In the present study, transgenic tobacco lines that express dsRNA molecules targeting a S. littoralis gene (i.e., Sl 102) were produced. The target gene is involved in major cellular immune responses (i.e., nodulation and encapsulation) in S. littoralis larvae (Di Lelio et al. 2014; Caccia et al. 2016). When larvae were fed transgenic tobacco lines, Sl 102 gene expression was reduced, and this reduction was reflected in the impairment of cellular immune responses in experimental larvae, as observed in our previous studies with naked and bacteria-delivered Sl 102 dsRNA (Di Lelio et al. 2014; Caccia et al. 2016, 2020). Additionally, silencing was influenced by the amount of plant tissue consumed by the larvae, confirming the results of previous studies. Indeed, feeding bioassays with plants expressing dsRNA demonstrated the importance of the amount or duration of food consumption to obtain significant gene silencing and relative phenotypic alterations in several insects (Baum et al. 2007; Zhu et al. 2012; Mao and Zheng 2014; Han et al. 2017; Hou et al. 2019).
Importantly, the present work showed that once silencing was effective, S. littoralis larvae were more susceptible to the Bt-based insecticide Xentari™ and this effect was observed in advanced instars. This issue is very relevant from an application point of view since Bt sprays are less active on older instar larvae that cause major damage to the crop due to increased feeding (Bryant 1994; Navon 2000). This evidence also corroborates our recently proposed model on the role of septicemia in Bt mode of action (Caccia et al. 2016), and in particular the importance of insect immune responses to counteract the microbial invasion of the hemocoel through midgut Bt-induced midgut lesions.
The strategy proposed herein would be extremely valuable to control pest populations with overlapping generations that require very high doses of Bt product, as high doses are necessary to kill older larvae but are completely unneeded for younger larvae (Bryant 1994; Navon 2000). In this context, immunosuppressive plants would minimize the dose of Bt products (i) to obtain high mortality in all larval stages and (ii) to reduce selective pressure and retard the development of resistance in susceptible insects. In fact, although less frequent than in Bt-crops, resistance outbreaks to Bt formulations should be considered a major concern for the long-term use of such an effective bioinsecticide (Tabashnik et al. 1990; Janmaat and Myers 2003; Hernández-Martínez et al. 2010; Zago et al. 2013; Jurat-Fuentes et al. 2021).
Immune suppressive RNAi-plants may therefore represent a significant component of future IPM strategies against lepidopteran larvae that involve MCA application. In conclusion, the present work provides a proof of concept for the exploitation of RNAi-plants that create conditions necessary to boost MCA effectiveness against poorly susceptible larval stages of lepidopteran pests rather than directly killing agricultural and horticultural lepidopteran pests.
Authors’ contributions
SC and RR conceived and designed research. EB, MC, SC, GC and IDL performed experiments. EB, MC, IDL and SC analyzed data. SC wrote the manuscript. All authors read and approved the manuscript.
References
Abdeen A, Virgos A, Olivella E, Villanueva J, Aviles X, Gabarra R, Prat S (2005) Multiple insect resistance in transgenic tomato plants over-expressing two families of plant proteinase inhibitors. Plant Mol Biol 57:189–202. https://doi.org/10.1007/s11103-004-6959-9
Abouseadaa HH, Osman GH, Ramadan AM, Hassanein SE, Abdelsattar MT, Morsy YB, Alameldin HF, El-Ghareeb DK, Nour-Eldin HA, Salem R, Gad AA, Elkhodary SE, Shehata MM, Mahfouz HM, Eissa HF, Bahieldin A (2015) Development of transgenic wheat (Triticum aestivum L.) expressing avidin gene conferring resistance to stored product insects. BMC Plant Biol 15:183. https://doi.org/10.1186/s12870-015-0570-x
Agrawal N, Dasaradhi PV, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK (2003) RNA interference: biology, mechanism, and applications. Microbiol Mol Biol Rev 67:657–685. https://doi.org/10.1128/mmbr.67.4.657-685.2003
Anderson JA, Ellsworth PC, Faria JC, Head GP, Owen MDK, Pilcher CD, Shelton AM, Meissle M (2019) Genetically engineered crops: importance of diversified integrated pest management for agricultural sustainability. Front Bioeng Biotechnol 7:24. https://doi.org/10.3389/fbioe.2019.00024
Barre A, Van Damme EJM, Simplicien M, Benoist H, Rougé P (2020) Are dietary lectins relevant allergens in plant food allergy? Foods 9:1724. https://doi.org/10.3390/foods9121724
Baum JA, Bogaert T, Clinton W, Heck GR, Feldmann P, Ilagan O, Johnson S, Plaetinck G, Munyikwa T, Pleau M, Vaughn T, Roberts J (2007) Control of coleopteran insect pests through RNA interference. Nat Biotechnol 25:1322–1326. https://doi.org/10.1038/nbt1359
Becchimanzi A, Di Lelio I, Pennacchio F, Caccia S (2019) Analysis of cellular immune response in Lepidopteran larvae. In: Sandrelli F, Tettamandi G (eds) Immunity in Insects. Humana, New York, pp 97–111
Berini F, Katz C, Gruzdev N, Casartelli M, Tettamanti G, Marinelli F (2018) Microbial and viral chitinases: attractive biopesticides for integrated pest management. Biotechnol Adv 36:818–838. https://doi.org/10.1016/j.biotechadv.2018.01.002
Boddupally D, Tamirisa S, Gundra SR, Vudem DR, Khareedu VR (2018) Expression of hybrid fusion protein (Cry1Ac::ASAL) in transgenic rice plants imparts resistance against multiple insect pests. Sci Rep 8:8458. https://doi.org/10.1038/s41598-018-26881-9
Boulter D, Edwards GA, Gatehouse AMR, Gatehouse JA, Hilder VA (1990) Additive protective effects of different plant-derived insect resistance genes in transgenic tobacco plants. Crop Protect 9:351–354. https://doi.org/10.1016/0261-2194(90)90005-R
Bryant JE (1994) Application strategies for Bacillus thuringiensis. Agric Ecosyst Environ 49:65–75
Burgess EPJ, Malone LA, Christeller JT, Lester MT, Murray C, Philip BA, Phung MM, Tregidga EL (2002) Avidin expressed in transgenic tobacco leaves confers resistance to two noctuid pests, Helicoverpa armigera and Spodoptera litura. Transgenic Res 11:185–198. https://doi.org/10.1023/a:1015297302990
Caccia S, Van Damme EJM, De Vos WH, Smagghe G (2012) Mechanism of entomotoxicity of the plant lectin from Hippeastrum hybrid (Amaryllis) in Spodoptera littoralis larvae. J Insect Physiol 58:1177–1183. https://doi.org/10.1016/j.jinsphys.2012.05.014
Caccia S, Di Lelio I, La Storia A, Marinelli A, Varricchio P, Franzetti E, Banyuls N, Tettamanti G, Casartelli M, Giordana B, Ferré J, Gigliotti S, Ercolini D, Pennacchio F (2016) Midgut microbiota and host immunocompetence underlie Bacillus thuringiensis killing mechanism. Proc Natl Acad Sci U S A 113:9486–9491. https://doi.org/10.1073/pnas.1521741113
Caccia S, Astarita F, Barra E, Di Lelio I, Varricchio P, Pennacchio F (2020) Enhancement of Bacillus thuringiensis toxicity by feeding Spodoptera littoralis larvae with bacteria expressing immune suppressive dsRNA. J Pest Sci 93:303–314. https://doi.org/10.1007/s10340-019-01140-6
Chen X, Li L, Hu Q, Zhang B, Wu W, Jin F, Jiang J (2015) Expression of dsRNA in recombinant Isaria fumosorosea strain targets the TLR7 gene in Bemisia tabaci. BMC Biotechnol 15:64. https://doi.org/10.1186/s12896-015-0170-8
Corrado G, Arciello S, Fanti P, Fiandra L, Garonna A, Digilio MC, Lorito M, Giordana B, Pennacchio F, Rao R (2008) The chitinase A from the baculovirus AcMNPV enhances resistance to both fungi and herbivorous pests in tobacco. Transgenic Res 17:557–571. https://doi.org/10.1007/s11248-007-9129-4
Corrado G, Alagna F, Rocco M, Renzone G, Varricchio P, Coppola V, Coppola M, Garonna A, Baldoni L, Scaloni A, Rao R (2012) Molecular interactions between the olive and the fruit fly Bactrocera oleae. BMC Plant Biol 12:86. https://doi.org/10.1186/1471-2229-12-86
Corrado G, Arena S, Araujo-Burgos T, Coppola M, Rocco M, Scaloni A, Rao R (2016) The expression of the tomato prosystemin in tobacco induces alterations irrespective of its functional domain. PCTOC 125:509–519. https://doi.org/10.1007/s11240-016-0967-8
Dani MP, Richards EH, Edwards JP (2004) Venom from the pupal endoparasitoid, Pimpla hypochondriaca, increases the susceptibility of larval Lacanobia oleracea to the entomopathogens Bacillus cereus and Beauveria bassiana. J Invertebr Pathol 86:19–25. https://doi.org/10.1016/j.jip.2004.03.007
Di Lelio I, Varricchio P, Di Prisco G, Marinelli A, Lasco V, Caccia S, Casartelli M, Giordana B, Rao R, Gigliotti S, Pennacchio F (2014) Functional analysis of an immune gene of Spodoptera littoralis by RNAi. J Insect Physiol 64:90–97. https://doi.org/10.1016/j.jinsphys.2014.03.008
Di Lelio I, Illiano A, Astarita F, Gianfranceschi L, Horner D, Varricchio P, Amoresano A, Pucci P, Pennacchio F, Caccia S (2019) Evolution of an insect immune barrier through horizontal gene transfer mediated by a parasitic wasp. PLoS Genet 15(3):e1007998. https://doi.org/10.1371/journal.pgen.1007998
Ding X, Gopalakrishnan B, Johnson LB, White FF, Wang X, Morgan TD, Kramer KJ, Muthukrishnan S (1998) Insect resistance of transgenic tobacco expressing an insect chitinase gene. Transgenic Res 7:77–84. https://doi.org/10.1023/a:1008820507262
Dormatey R, Sun C, Ali K, Coulter JA, Bi Z, Bai J (2020) Gene pyramiding for sustainable crop improvement against biotic and abiotic stresses. Agronomy 10:1255. https://doi.org/10.3390/agronomy10091255
Dunaevsky YE, Elpidina EN, Vinokurov KS, Belozersky MA (2005) Protease inhibitors in improvement of plant resistance to pathogens and insects. Mol Biol 39:608–613. https://doi.org/10.1007/s11008-005-0076-y
Eakteiman G, Moses-Koch R, Moshitzky P, Mestre-Rincon N, Vassão DG, Luck K, Sertchook R, Malka O, Morin S (2018) Targeting detoxification genes by phloem-mediated RNAi: a new approach for controlling phloem-feeding insect pests. Insect Biochem Mol Biol 100:10–21. https://doi.org/10.1016/j.ibmb.2018.05.008
Fan X, Shi X, Zhao J, Zhao R, Fan Y (1999) Insecticidal activity of transgenic tobacco plants expressing both Bt and CpTI genes on cotton bollworm (Helicoverpa armigera). Chin J Biotechnol 15:1–5
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811. https://doi.org/10.1038/35888 (PMID: 9486653)
Fitches EC, Pyati P, King GF, Gatehouse JA (2012) Fusion to snowdrop lectin magnifies the oral activity of insecticidal ω-hexatoxin-Hv1a peptide by enabling its delivery to the central nervous system. PLoS ONE 7(6):e39389. https://doi.org/10.1371/journal.pone.0039389
Fletcher SJ, Reeves PT, Hoang BT, Mitter N (2020) A perspective on RNAi-based biopesticides. Front Plant Sci 11:51. https://doi.org/10.3389/fpls.2020.00051
Foissac X, Loc NT, Christou P, Gatehouse AMR, Gatehouse JA (2000) Resistance to green leafhopper (Nephotettix virescens) and brown planthopper (Nilaparvata lugens) in transgenic rice expressing snowdrop lectin (Galanthus nivalis agglutinin; GNA). J Insect Physiol 46:573–583. https://doi.org/10.1016/s0022-1910(99)00143-2
Fu S, Liu Z, Chen J, Sun G, Jiang Y, Li M, Xiong L, Chen S, Zhou Y, Asad M, Yang G (2020) Silencing arginine kinase/integrin β1 subunit by transgenic plant expressing dsRNA inhibits the development and survival of Plutella xylostella. Pest Manag Sci 76:1761–1771. https://doi.org/10.1002/ps.5701
Fulton TM, Chunwongse J, Tanksley SD (1995) Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol Biol 13:207–209. https://doi.org/10.1007/BF02670897
Gasmi L, Boulain H, Gauthier J, Hua-Van A, Musset K, Jakubowska AK, Aury JM, Volkoff AN, Huguet E, Herrero S, Drezen JM (2015) Recurrent domestication by Lepidoptera of genes from their parasites mediated by Bracoviruses. PLoS Genet 11(9):e1005470. https://doi.org/10.1371/journal.pgen.1005470
Gatehouse AM, Ferry N, Edwards MG, Bell HA (2011) Insect-resistant biotech crops and their impacts on beneficial arthropods. Philos Trans R Soc Lond B Biol Sci 366:1438–1452. https://doi.org/10.1098/rstb.2010.0330
Ghildiyal M, Zamore PD (2009) Small silencing RNAs: an expanding universe. Nat Rev Genet 10:94–108. https://doi.org/10.1038/nrg2504
Guo W, Bai C, Wang Z, Wang P, Fan Q, Mi X, Wang L, He J, Pang J, Luo X, Fu W, Tian Y, Si H, Zhang G, Wu J (2018) Double-Stranded RNAs high-efficiently protect transgenic potato from Leptinotarsa decemlineata by disrupting juvenile hormone biosynthesis. J Agric Food Chem 66:11990–11999. https://doi.org/10.1021/acs.jafc.8b03914
Han Q, Wang Z, He Y, Xiong Y, Lv S, Li S, Zhang Z, Qiu D, Zeng H (2017) Transgenic cotton plants expressing the HaHR3 gene conferred enhanced resistance to Helicoverpa armigera and improved cotton yield. Int J Mol Sci 18:1874. https://doi.org/10.3390/ijms18091874
Helliwell CA, Wesley SV, Wielopolska AJ, Waterhouse P (2002) High-throughput vectors for efficient gene silencing in plants. Funct Plant Biol 29:1217–1225. https://doi.org/10.1071/FP02033
Hernández-Martínez P, Navarro-Cerrillo G, Caccia S, de Maagd RA, Moar WJ, Ferré J, Escriche B, Herrero S (2010) Constitutive activation of the midgut response to Bacillus thuringiensis in Bt-resistant Spodoptera exigua. PLoS ONE 5(9):e12795. https://doi.org/10.1371/journal.pone.0012795
Hou Q, Xu L, Liu G, Pang X, Wang X, Zhang Y, You M, Ni Z, Zhao Z, Liang R (2019) Plant-mediated gene silencing of an essential olfactory-related Gqα gene enhances resistance to grain aphid in common wheat in greenhouse and field. Pest Manag Sci 75:1718–1725. https://doi.org/10.1002/ps.5292
Janmaat AF, Myers J (2003) Rapid evolution and the cost of resistance to Bacillus thuringiensis in greenhouse populations of cabbage loopers, Trichoplusia ni. Proc Biol Sci 270:2263–2270. https://doi.org/10.1098/rspb.2003.2497
Jurat-Fuentes JL, Heckel DG, Ferré J (2021) Mechanisms of resistance to insecticidal proteins from Bacillus thuringiensis. Annu Rev Entomol 66:121–140. https://doi.org/10.1146/annurev-ento-052620-073348
Kim D, Rossi J (2008) RNAi mechanisms and applications. Biotechniques 44:613–616. https://doi.org/10.2144/000112792
Kramer KJ, Morgan TD, Throne JE, Dowell FE, Bailey M, Howard JA (2000) Transgenic avidin maize is resistant to storage insect pests. Nat Biotechnol 18:670–674. https://doi.org/10.1038/76531
Liu SM, Li J, Zhu JQ, Wang XW, Wang CS, Liu SS, Chen XX, Li S (2016) Transgenic plants expressing the AaIT/GNA fusion protein show increased resistance and toxicity to both chewing and sucking pests. Insect Sci 23:265–276. https://doi.org/10.1111/1744-7917.12203
Liu S, Jaouannet M, Dempsey DA, Imani J, Coustau C, Kogel KH (2020) RNA-based technologies for insect control in plant production. Biotechnol Adv 39:107463. https://doi.org/10.1016/j.biotechadv.2019.107463
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Macedo MLR, Oliveira CFR, Oliveira CT (2015) Insecticidal activity of plant lectins and potential application in crop protection. Molecules 20:2014–2033. https://doi.org/10.3390/molecules20022014
Mamta B, Rajam MV (2017) RNAi technology: a new platform for crop pest control. Physiol Mol Biol Plants 23:487–501. https://doi.org/10.1007/s12298-017-0443-x
Mao J, Zeng F (2014) Plant-mediated RNAi of a gap gene-enhanced tobacco tolerance against the Myzus persicae. Transgenic Res 23:145–152. https://doi.org/10.1007/s11248-013-9739-y
Mao YB, Cai WJ, Wang JW, Hong GJ, Tao XY, Wang LJ, Huang YP, Chen XY (2007) Silencing a cotton bollworm P450 monooxygenase gene by plant-mediated RNAi impairs larval tolerance of gossypol. Nat Biotechnol 25:1307–1313. https://doi.org/10.1038/nbt1352
Mao YB, Tao XY, Xue XY, Wang LJ, Chen XY (2011) Cotton plants expressing CYP6AE14 double-stranded RNA show enhanced resistance to bollworms. Transgenic Res 20:665–673. https://doi.org/10.1007/s11248-010-9450-1
Maqbool SB, Riazzudin S, Loc NT, Gatehouse AMR, Gatehouse JA, Christou P (2001) Expression of multiple insecticidal genes confers broad resistance against a range of different rice pests. Mol Breed 7:85–93. https://doi.org/10.1023/A:1009644712157
Mi X, Liu X, Yan H, Liang L, Zhou X, Yang J, Si H, Zhang N (2017) Expression of the Galanthus nivalis agglutinin (GNA) gene in transgenic potato plants confers resistance to aphids. C R Biol 340:7–12. https://doi.org/10.1016/j.crvi.2016.10.003
Moreira-Pinto CE, Ramos Coelho R, Borges Leite AG, Amaral Silveira D, Aguiar Souza D, Biaggioni Lopes R, Lima Pepino Macedo L, Mattar Silva MC, Pires Ribeiro T, Morgante CV, Antonino JD, Grossi-de-Sa MF (2021) Increasing Anthonomus grandis susceptibility to Metarhizium anisopliae through RNAi-induced AgraRelish knockdown: a perspective to combine biocontrol and biotechnology. Pest Manag Sci 77:4054–4063. https://doi.org/10.1002/ps.6430
Nagadhara D, Ramesh S, Pasalu IC, Rao YK, Sarma NP, Reddy VD, Rao KV (2004) Transgenic rice plants expressing the snowdrop lectin gene (gna) exhibit high-level resistance to the whitebacked planthopper (Sogatella furcifera). Theor Appl Genet 109:1399–1405. https://doi.org/10.1007/s00122-004-1750-5
Nakasu EY, Edwards MG, Fitches E, Gatehouse JA, Gatehouse AM (2014) Transgenic plants expressing ω-ACTX-Hv1a and snowdrop lectin (GNA) fusion protein show enhanced resistance to aphids. Front Plant Sci 5:673. https://doi.org/10.3389/fpls.2014.00673
Navon (2000) Bacillus thuringiensis insecticides in crop protection-reality and prospects. Crop Prot 19:669–676. https://doi.org/10.1016/S0261-2194(00)00089-2
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:2002–2007. https://doi.org/10.1093/nar/29.9.e45
Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST(C)) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30:e36. https://doi.org/10.1093/nar/30.9.e36
Rao KV, Rathore KS, Hodges TK, Fu X, Stoger E, Sudhakar D, Williams S, Christou P, Bharathi M, Bown DP, Powell KS, Spence J, Gatehouse AM, Gatehouse JA (1998) Expression of snowdrop lectin (GNA) in transgenic rice plants confers resistance to rice brown planthopper. Plant J 15:469–477. https://doi.org/10.1046/j.1365-313X.1998.00226.x
Rauf I, Javaid S, Naqvi RZ, Mustafa T, Amin I, Mukhtar Z, Jander G, Mansoor S (2019) In-planta expression of insecticidal proteins provides protection against lepidopteran insects. Sci Rep 9:6745. https://doi.org/10.1038/s41598-019-41833-7
Richards EH, Dani MP (2010) A recombinant immunosuppressive protein from Pimpla hypochondriaca (rVPr1) increases the susceptibility of Lacanobia oleracea and Mamestra brassicae larvae to Bacillus thuringiensis. J Invertebr Pathol 104:51–57. https://doi.org/10.1016/j.jip.2010.01.010
Richards EH, Bradish H, Dani MP, Pietravalle S, Lawson A (2011) Recombinant immunosuppressive protein from Pimpla hypochondrica venom (rVPr1) increases the susceptibility of Mamestra brassicae larvae to the fungal biological control agent, Beauveria bassiana. Arch Insect Biochem Physiol 78:119–131. https://doi.org/10.1002/arch.20447
Richards EH, Dani MP, Bradish H (2013) Immunosuppressive properties of a protein (rVPr1) from the venom of the endoparasitic wasp, Pimpla hypochondriaca: Mechanism of action and potential use for improving biological control strategies. J Insect Physiol 59:213–222. https://doi.org/10.1016/j.jinsphys.2012.06.001
Ricroch AE, Hénard-Damave MC (2016) Next biotech plants: new traits, crops, developers and technologies for addressing global challenges. Criti Rev Biotechnol 36:675–690. https://doi.org/10.3109/07388551.2015.1004521
Rivkin H, Kroemer JA, Bronshtein A, Belausov E, Webb BA, Chejanovsky N (2006) Response of immunocompetent and immunosuppressed Spodoptera littoralis larvae to baculovirus infection. J Gen Virol 87:2217–2225. https://doi.org/10.1099/vir.0.81918-0
Sainsbury F, Benchabane M, Goulet MC, Michaud D (2012) Multimodal protein constructs for herbivore insect control. Toxins 4:455–475. https://doi.org/10.3390/toxins4060455
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harb Lab Press, New York.
Schlüter U, Benchabane M, Munger A, Kiggundu A, Vorster J, Goulet MC, Cloutier C, Michaud D (2010) Recombinant protease inhibitors for herbivore pest control: a multitrophic perspective. J Exp Bot 61:4169–4183. https://doi.org/10.1093/jxb/erq166
Schuster S, Miesen P, van Rij RP (2019) Antiviral RNAi in insects and mammals: parallels and differences. Viruses 11:448. https://doi.org/10.3390/v11050448
Senthilkumar R, Cheng CP, Yeh KW (2010) Genetically pyramiding protease-inhibitor genes for dual braod-spectrum resistance against insect and phytopathogens in transgenic tobacco. Plant Biotechnol J 8:65–75. https://doi.org/10.1111/j.1467-7652.2009.00466.x
Sétamou M, Bernal JS, Legaspi JC, Mirkov TE, Legaspi JC Jr (2002) Evaluation of lectin-expressing transgenic sugarcane against stalkborers (Lepidoptera: Pyralidae): effects on life history parameters. J Econ Entomol 95:469–477. https://doi.org/10.1603/0022-0493-95.2.469
Shahidi-Noghabi S, Van Damme EJ, Mahdian K, Smagghe G (2010a) Entomotoxic action of Sambucus nigra agglutinin I in Acyrthosiphon pisum aphids and Spodoptera exigua caterpillars through caspase-3-like-dependent apoptosis. Arch Insect Biochem Physiol 75:207–220. https://doi.org/10.1002/arch.20387
Shahidi-Noghabi S, Van Damme EJ, Iga M, Smagghe G (2010b) Exposure of insect midgut cells to Sambucus nigra L. agglutinins I and II causes cell death via caspase-dependent apoptosis. J Insect Physiol 56:1101–1107. https://doi.org/10.1016/j.jinsphys.2010.03.012
Singh S, Singh A, Kumar S, Mittal P, Singh IK (2020) Protease inhibitors: recent advancement in its usage as a potential biocontrol agent for insect pest management. Insect Sci 27:186–201. https://doi.org/10.1111/1744-7917.12641
Su X, Chu Y, Li H, Hou Y, Zhang B, Huang Q, Hu Z, Huang R, Tian Y (2011) Expression of multiple resistance genes enhances tolerance to environmental stressors in transgenic poplar (Populus×euramericana ‘Guariento’). PLoS ONE 6(9):e24614. https://doi.org/10.1371/journal.pone.0024614
Tabashnik BE, Cushing NL, Finson N, Johnson MA (1990) Field development of resistance to Bacillus thuringiensis in diamondback moth (Lepidoptera: Plutellidae). J Econ Entomol 83:1671–1676. https://doi.org/10.1093/jee/83.5.1671
Trung NP, Fitches E, Gatehouse JA (2006) A fusion protein containing a lepidopteran-specific toxin from the South Indian red scorpion (Mesobuthus tamulus) and snowdrop lectin shows oral toxicity to target insects. BMC Biotech 6:18. https://doi.org/10.1186/1472-6750-6-18
Vandenborre G, Smagghe G, Van Damme EJM (2011) Plant lectins as defense proteins against phytophagous insects. Phytochemistry 72:1538–1550. https://doi.org/10.1016/j.phytochem.2011.02.024
Vasconcelos IM, Oliveira JTA (2004) Antinutritional properties of plant lectins. Toxicon 44:385–403. https://doi.org/10.1016/j.toxicon.2004.05.005
Wang X, Ding X, Gopalakrishnan B, Morgan TD, Johnson L, White FF, Muthukrishnan S, Kramer KJ (1996) Characterization of a 46 kDa insect chitinase from transgenic tobacco. Insect Biochem Mol Biol 26:1055–1064. https://doi.org/10.1016/S0965-1748(96)00056-2
Wang Z, Zhang K, Sun X, Tang K, Zhang J (2005) Enhancement of resistance to aphids by introducing the snowdrop lectin gene gna into maize plants. J Biosci 30:627–638. https://doi.org/10.1007/BF02703563
Washburn JO, Haas-Stapleton EJ, Tan FF, Beckage NE, Volkman LE (2000) Co-infection of Manduca sexta larvae with polydnavirus from Cotesia congregata increases susceptibility to fatal infection by Autographa californica M Nucleopolyhedrovirus. J Insect Physiol 46:179–190. https://doi.org/10.1016/S0022-1910(99)00115-8
Zago HB, Siqueira HÁ, Pereira EJ, Picanço MC, Barros R (2013) Resistance and behavioural response of Plutella xylostella (Lepidoptera: Plutellidae) populations to Bacillus thuringiensis formulations. Pest Manag Sci 70:488–495. https://doi.org/10.1002/ps.3600
Zhang B, Chen M, Zhang X, Luan H, Diao S, Tian Y, Su X (2011) Laboratory and field evaluation of the transgenic Populus alba×Populus glandulosa expressing double coleopteran-resistance genes. Tree Physiol 31:567–573. https://doi.org/10.1093/treephys/tpr032
Zhang J, Khan SA, Hasse C, Ruf S, Heckel DG, Bock R (2015) Full crop protection from an insect pest by expression of long double-stranded RNAs in plastids. Science 347:991–994. https://doi.org/10.1126/science.1261680
Zhang J, Khan SA, Heckel DG, Bock R (2017) Next-generation insect-resistant plants: RNAi-mediated crop protection. Trends Biotechnol 35:871–882. https://doi.org/10.1016/j.tibtech.2017.04.009
Zhu KY, Palli SR (2020) Mechanisms, applications, and challenges of insect RNA interference. Annu Rev Entomol 65:293–311. https://doi.org/10.1146/annurev-ento-011019-025224
Zhu JQ, Liu S, Ma Y, Zhang JQ, Qi HS, Wei ZJ, Yao Q, Zhang WQ, Li S (2012) Improvement of pest resistance in transgenic tobacco plants expressing dsRNA of an insect-associated gene EcR. PLoS ONE 7(6):e38572. https://doi.org/10.1371/journal.pone.0038572
Zhu-Salzman K, Zeng R (2015) Insect response to plant defensive protease inhibitors. Annu Rev Entomol 60:233–252. https://doi.org/10.1146/annurev-ento-010814-020816
Funding
This work was supported by the Ministero dell’Istruzione, dell’Università e della Ricerca, Futuro in Ricerca 2013 (RBFR13PMT1) (to SC) and by the European Union’s Horizon 2020 research and innovation program, under Grant Agreement No. 773554 (EcoStack).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest/competing interests.
Additional information
Communicated by Subba Reddy Palli.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Di Lelio, I., Barra, E., Coppola, M. et al. Transgenic plants expressing immunosuppressive dsRNA improve entomopathogen efficacy against Spodoptera littoralis larvae. J Pest Sci 95, 1413–1428 (2022). https://doi.org/10.1007/s10340-021-01467-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10340-021-01467-z