
Abstract
Although COVID-19 presents primarily as a lower respiratory tract infection transmitted via air droplets, increasing data suggest multiorgan involvement in patients that are infected. This systemic involvement is postulated to be mainly related to the SARS-CoV-2 virus binding on angiotensin-converting enzyme 2 (ACE2) receptors located on several different human cells. Lung involvement is the most common serious manifestation of the disease, ranging from asymptomatic disease or mild pneumonia, to severe disease associated with hypoxia, critical disease associated with shock, respiratory failure and multiorgan failure or death. Among patients with COVID-19, underlying cardiovascular comorbidities including hypertension, diabetes and especially cardiovascular disease, has been associated with adverse outcomes, whereas the emergence of cardiovascular complications, including myocardial injury, heart failure and arrhythmias, has been associated with poor survival. Gastrointestinal symptoms are also frequently encountered and may persist for several days. Haematological complications are frequent as well and have been associated with poor prognosis. Furthermore, recent studies have reported that over a third of infected patients develop a broad spectrum of neurological symptoms affecting the central nervous system, peripheral nervous system and skeletal muscles, including anosmia and ageusia. The skin, the kidneys, the liver, the endocrine organs and the eyes are also affected by the systemic COVID-19 disease. Herein, we provide a comprehensive overview of the organ-specific systemic manifestations of COVID-19.
Similar content being viewed by others

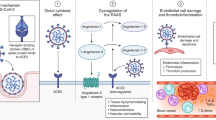
Introduction
The SARS-CoV-2 virus has caused a worldwide pandemic in the past few months with a major impact on health care systems and economies. Since December 2019 when the first diagnosed case was identified in Wuhan, China, the highly contagious virus has spread throughout the world with detrimental consequences. SARS-CoV-2 belongs to the coronaviruses family of enveloped, single-stranded RNA viruses [1]. Notably, the main hosts of these viruses are animals. To date, 39 different species of the viruses have been identified, including two highly contagious and pathogenic species that led to two different outbreaks the past 2 decades [severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002 and Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012] [2]. Patients infected with this new coronavirus present with a variety of symptoms, which range from asymptomatic disease to mild and moderate symptoms (mild pneumonia), severe symptoms (dyspnoea, hypoxia, or > 50% lung involvement on imaging) and symptoms of critical illness (acute respiratory distress syndrome, respiratory failure, shock or multiorgan system dysfunction). The disease affects mainly elderly adults; however, younger patients without comorbidities can also be diagnosed with severe disease. The virus presents primarily as a lower tract respiratory infection transmitted via air droplets, but the multisystemic nature of the disease is becoming increasingly apparent as more data are emerging. It is postulated that it is related to the tropism of the virus for the ACE-2 receptors located on several different human cells. The occurrence of other symptoms can not only coexist, but may also precede the typical phenotype of COVID-19. In a recent study, SARS-CoV-2 viral load was quantified in 22 post-mortem autopsy tissue samples [3]; 17 patients (77%) had more than two coexisting conditions. The number of coexisting conditions was strongly associated with SARS-CoV-2 affinity to the kidneys, including patients without history of chronic kidney disease. The highest levels of SARS-CoV-2 copies were detected in the respiratory tract, while the levels detected in kidneys, liver, heart, brain and blood were lower. These findings indicate a possible organ tropism of SARS-CoV-2 that might influence the course of the disease leading potentially to underlying conditions aggravation. As our knowledge on the virus mechanisms increases, our understanding on the various complications will continue to evolve. This manuscript aims to review the available literature and provide further insight on multiorgan involvement of the disease (Fig. 1).

Schematic overview of the systemic manifestations of COVID-19 infection and the underlying pathophysiology
Pulmonary involvement
The severity of lung involvement associated with SARS-CoV-2 infection ranges from lack of symptoms or mild pneumonia (in 81%) to severe disease-associated hypoxia (seen in 14%), critical disease associated with shock, respiratory failure and multiorgan failure (in 5%) or death (2.3%) [4]. It is the most common serious disease manifestation. Patients may present with dry cough, fever, sputum production, fatigue and dyspnea, and the reported frequency varies based on the cohort studied [5,6,7]. Among hospitalized patients, 20–41% will develop acute respiratory distress syndrome (ARDS). What is becoming increasingly apparent as our understanding of the mechanisms of COVID-19 induced lung injury expands, are the distinct or “atypical” features of COVID-19-associated ARDS [4, 8].
Based on SARS-CoV virus data, the genome of which is highly homologous to the SARS-CoV-2 genome, it is hypothesized that the human angiotensin-converting enzyme 2 (ACE2) receptor is the main functional receptor for the SARS-CoV-2 virus. The ACE2 receptor is expressed on the apical side of type II alveolar epithelial cells in the alveolar space, and the large surface area of the lung serves as a reservoir for viral binding and replication, providing an explanation for the tropism of the SARS-CoV-2 virus and the lung vulnerability observed [9, 10].
SARS-CoV-2 infection induces alveolar injury and interstitial inflammation. Dendritic cells (DCs) and alveolar macrophages phagocytose the virus-infected apoptosed epithelial cells, and T cell responses are initiated activating innate and adaptive immune mechanisms [11]. Levels of proinflammatory cytokines and chemokines, such as tumour necrosis factor (TNF)-α, interleukin 1β (IL-1β), IL-6, and more, are increased in patients with COVID-19 infection [12]. The cytokine storm is hypothesized to play a central role in the immunopathology of COVID-19, but the primary source or the exact virological mechanisms behind it have not been identified yet. There is extensive hemophagocytosis which shares features but is distinct from the well-described macrophage activation syndrome (MAS) [13, 14]. In addition to the proinflammatory stage and immune system activation, an immune suppression stage follows which is characterized by lymphopenia, low CD4 and CD8 T cell counts, increasing the risk of bacterial infection [15, 16].
Emerging data from COVID-19 pneumonia autopsy studies demonstrate acute interstitial pneumonia and diffuse alveolar damage (DAD) with macrophage infiltration, formation of hyaline membranes and alveolar wall oedema and thickening. There is also microvasculature involvement with pulmonary vessel (intra and extra) hyaline thrombosis, haemorrhage, vessel wall oedema, intravascular neutrophil trapping and immune cell infiltration. In one series, in 5 out of 23 patients, major pulmonary vessel thromboemboli and/or haemorrhage were reported [14, 17, 18]. McGonagle et al. use the term diffuse pulmonary intravascular coagulopathy (PIC) to describe this lung-restricted vascular immunopathology [19] driven probably by the close anatomical positioning of type II pneumocytes and the pulmonary vasculature. At early stages of this process, there is no systemic coagulopathy (DIC) which is seen, however, at later disease stages coupled with the presence of ARDS. Extensive microthrombi formation within the vascular bed causes pulmonary infarction, haemorrhage, pulmonary hypertension and secondary ventricular stress [20]. Hypoxemia and mechanical ventilation which forces immunostimulatory molecules in the microvasculature also seem to contribute to the development of PIC.
Findings on chest radiograph imaging are not disease-specific and usually include ground glass opacities with bilateral, peripheral or lower lung zone distribution with or without consolidation [21,22,23]. Chest CT is more sensitive, but no finding can 100% establish or rule out the diagnosis [24]. According to the Radiological Society of North America, CT findings are categorized into typical, indeterminate or atypical for COVID-19 demonstrating that specificity is low even for CT [25]. In one study using RT-PCR as a reference, sensitivity was 97% but specificity very low at 25% [26]. Radiological abnormalities increase over the disease course, and the typical peak is at 10–12 days post-symptom initiation. At early stages or in mild disease, imaging may not reveal any pathology, but interestingly abnormal findings on imaging can be identified in some cases prior to symptom development or even prior to PCR RNA detection [27]. Using CT as a screening tool is not, however, recommended. Improvement of the findings lags behind symptom or hypoxia improvement [28].
Hypoxia is frequently a presenting feature of COVID-19 pneumonia, but interestingly, it is often insidious and paradoxically well tolerated by the patients. This unusual clinical presentation, seen at early disease stages, is referred to as “silent hypoxia” and is linked to the “atypical” features of the ARDS syndrome associated with COVID-19 pneumonia [29].
Contrary to the typical ARDS, lung compliance is preserved and the hypoxia-driven tachypnea allows high volumes and hypocapnia which fails to stimulate the sensation of dyspnea. A similar pathophysiological mechanism is seen in hypobaric hypoxia at high altitude [30]. A model has been recently proposed which includes two time-associated phenotypes. The severity of infection, patient comorbidities and physiological reserve, the time elapsed between disease onset and presentation to hospital and the host immune response all contribute. The L-phenotype is seen at early disease stages; there is high lung compliance, and the ventilation-to-perfusion ratio (Va/Q ratio) is low, but there is dysfunctional regulation of perfusion with hypoxic vasoconstriction. At this stage, the lung weight is low and lung recruitability is low with minimal amount of non-aerated lung tissue. This develops into the H-phenotype with decreased lung compliance due to oedema, increased fraction of cardiac output perfusing the non-aerated tissue and therefore a right-to-left shunt, increased lung volume due to oedema and consolidation and therefore high recruitability. Type L patients usually remain stable for some time and can then either improve or worsen and transition into type H, secondary to evolution of the COVID-19 pneumonia but also injury induced by high-stress ventilation [31].
Management should be adapted based on the type of phenotype and timing/stage of lung injury. A high positive end-expiratory pressure (PEEP) on the ventilator, at early stages of poor lung recruitability, is not very effective, but in combination with gravitational forces (using prone positioning), it may allow for perfusion redistribution and increased oxygenation [32, 33]. The initial practice of early intubation was not supported by emerging data. L-phenotype patients should receive high-flow nasal cannula FiO2, continuous positive airway pressure or non-invasive ventilation with close monitoring. Awake or self-proning has been incorporated in many hospital protocols in an attempt to prevent intubation and reverse hypoxemia [34]. Type H patients should be treated as severe ARDS with higher PEEP volumes, prone positioning and extracorporeal support.
Another factor that may predispose for severe and potentially fatal ARDS in patients with COVID-19 is the excessive increase in circulating proinflammatory cytokines including interleukins (IL-1, IL-6), interferon and TNF-α. This “cytokine storm” results from an inflammatory over-reaction as a response to SARS-CoV-2 infection that ultimately leads to endothelial cell dysfunction, damage of the vascular barrier, capillary leak and diffuse alveolar damage [35]. In this context, anti-IL-6 inhibitors, such as the monoclonal antibody tocilizumab, inhibitors of JAK kinases, such as baricitinib, and corticosteroids, especially dexamethasone, have been evaluated in patients with severe COVID-19 and have shown promising preliminary results [36,37,38,39].
Cardiovascular manifestations
Among patients with COVID-19, underlying cardiovascular comorbidities including hypertension, diabetes, and especially cardiovascular disease, have been associated with adverse outcomes [4, 40,41,42,43,44,45,46,47,48,49], whereas the emergence of cardiovascular complications, including myocardial injury, heart failure and arrhythmias, has been associated with poor survival [6, 40,41,42,43,44, 50,51,52,53,54]. The presence of obesity is also associated with adverse cardiovascular outcomes [55].
Evidence of myocardial injury in patients with COVID-19 has been a remarkable finding [40,41,42, 44, 50,51,52,53]. ACE2 expression is significantly elevated in cardiac tissue [56] and may potentially facilitate direct myocardial damage induced by viral infection. There have been also reported isolated cases of COVID-19-induced myocarditis, which support the hypothesis of direct myocardial injury by SARS-CoV-2 [57,58,59,60]. Furthermore, ACE2 plays an important role in the renin–angiotensin system by catalysing the conversion of angiotensin II to angiotensin 1–7, which exerts a protective effect on the cardiovascular system [61, 62]. Importantly, the binding of SARS-CoV-2 to ACE2 is anticipated to result in loss of the external ACE2 catalytic effect [63, 64]. Subsequently, the theoretical downregulation of ACE2 and the decrease in angiotensin 1-7 levels in patients with COVID-19 may also compromise heart function [65].
In addition to the above, a non-negligible proportion of patients with COVID-19 seem to experience a hyperinflammatory state, in which inflammatory cytokines and other markers of systemic inflammation are markedly increased [40,41,42, 66]. The circulating cytokines can stimulate macrophages and leucocyte adhesion molecule expression on the endothelial cells of underlying atherosclerotic lesions, rendering them more vulnerable for disruption and increasing the possibility of a clinically evident acute coronary syndrome [67, 68]. Systemic cytokines may also activate the microvascular endothelium and induce a dysfunction of the coronary microvasculature, which may result in myocardial ischaemia and myocardial injury [68]. Inflammation and subsequent dysfunction of the endothelium in several organs are the result of both the direct effect of SARS-CoV-2 infection of endothelial cells and the indirect effects of the host inflammatory response [69].
Myocardial injury can also result secondary to a mismatch between myocardial oxygen supply and demand, known as type 2 myocardial infarction. SARS-CoV-2 infection may be associated with myocardial damage through increased myocardial oxygen demand along with reduced myocardial oxygen supply. Severe respiratory complications and associated hypoxia have been common findings in patients with COVID-19 [41, 43, 45, 51, 70]. Moreover, hypotension, which is a common clinical feature both in sepsis and during the cytokine storm syndrome, can also reduce myocardial oxygen supply [68]. Furthermore, systemic infection and fever increase the metabolic needs of peripheral tissues and end-organs, which elevates the metabolic demands of the myocardial cells [71].
Fulminant myocarditis may be a clinical manifestation of COVID-19 [57, 58] and may result in left ventricular systolic dysfunction or even cardiogenic shock [72, 73]. Among 176 Chinese patients with COVID-19, Chen et al. reported heart failure as a complication in 24.4% (n = 43), using age-related amino-terminal pro-brain natriuretic peptide (NTproBNP) cut-points [53]. Interestingly, there was a significant difference in the prevalence of heart failure between COVID-19 survivors and non-survivors (3.2% vs. 49.4%) [53]. Another study encompassing data from 191 patients reported a 23% (n = 44) incidence of heart failure, 63.6% (n = 28) of which was fatal [41]. A meta-analysis of 43 studies involving 3600 patients reported a prevalence of heart failure as a COVID-19 complication of 17.1% (95%, CI 1.5–42.2) among critically ill patients compared to 1.9% (95% CI 0.0–26.0) among non-critically ill patients [54].
Guo et al. reported sustained ventricular tachycardia or ventricular fibrillation in 5.9% (n = 11) of 187 patients treated in a COVID-19 specialized centre in China [52]. Another retrospective study including data from the 393 consecutive patients with COVID-19 in two hospitals in New York City showed that patients who received mechanical ventilation were more likely to have atrial arrhythmias (18.5% versus 1.9%) [6]. It has to be noted that sustained ventricular arrhythmias have been reported as a frequent clinical feature of acute myocarditis [73], which may be the case in patients with COVID-19 complicated by myocarditis. Importantly, arrhythmias may be also induced by medical treatment for COVID-19, such as chloroquine phosphate, hydroxychloroquine sulphate and azithromycin [74]. These agents and their combinations may prolong the QTc interval and predispose for torsades de pointes or other ventricular arrhythmias [75].
Another characteristic manifestation of COVID-19 in the cardiovascular system that has been recently reported is a Kawasaki-like syndrome, which is characterized by circulatory dysfunction and macrophage activation syndrome [76]. A single-centre study conducted in Bergamo, Italy, reported a 30-fold increase in the incidence of Kawasaki-like disease during the COVID-19 pandemic, as compared with the previous year [77]. The cytokine storm associated with infection by SARS-CoV-2 may be the predisposing mechanism for the Kawasaki-like clinical phenotype, but further insight has to be shed by future preclinical studies.
Gastrointestinal and liver involvement
The underlying pathophysiologic mechanism for the occurrence of digestive symptoms is also thought to be related to the virus’s affinity for ACE2 receptors located in specific enterocytes in the ileum and colon [52, 78, 79]. ACE2 receptors are involved partially in inflammation mechanisms and therefore could provide an explanation for the occurrence of diarrhoea in infected patients. Importantly, the binding efficiency is stronger for SARS-CoV-2 than the SARS-CoV-1, and this might be one of the reasons of high rate of transmission [80]. Binding to primary intestinal epithelial cells also raises the question on whether the virus can be transmitted through the faecal–oral route, which currently remains unconfirmed [78, 79]. The largest study evaluating digestive involvement in patients with COVID-19 was performed in Wuhan, China. The investigators evaluated 1141 retrospective cases admitted to one single hospital over a period of 7 weeks [81]. 16% (183) of patients presented only with gastrointestinal symptoms. The most common symptom reported was loss of appetite. Vomiting and nausea occurred in approximately two-third of the patients, while diarrhoea and abdominal pain were present in 37% and 25%, respectively. The main study limitations were its retrospective design along with the relatively small sample size. Another cross-sectional study from Hubei province reported results on digestive symptoms from patients being admitted to one of three different hospitals during January and February 2020. In total, 99 patients (48.5%) had gastrointestinal symptoms. The symptoms included anorexia (83.8%), diarrhoea (29.3%), vomiting (8.1%) and abdominal pain (4.0%), while some patients reported symptoms combinations. Notably, 7 patients presented only with digestive symptoms with no evidence of respiratory involvement. In this case, the diagnosis was delayed due to the non-specific symptoms they experienced [82]. Another study in China included 1099 patients and demonstrated that the most common symptoms on admission were fever (43.8%) and cough (67.8%) [43]. Gastrointestinal symptoms were less common—nausea or vomiting 5% and diarrhoea 3.8%, respectively. In a single-centre case series of 138 hospitalized patients with COVID-19, 10.1% reported diarrhoea and/or nausea, but the proportion of patients only with digestive symptoms was not outlined [50]. In another recent study performed in China, gastrointestinal symptoms were reported in 74 of 651 (11.4%) patients [83]. Nausea, vomiting and diarrhoea were the most common. Importantly, it was demonstrated that GI symptoms were more common in patients with severe COVID-19 disease (23% vs. 8.1%). Further data are required to understand better the role of the gastrointestinal involvement of COVID-19 and clarify whether it is correlated with worse outcomes. The abovementioned studies did not test for virus RNA in the stool, so there is no proof that active viral RNA replication can be found in the digestive tract. A recent report of a 25-year-old female who presented with respiratory symptoms and fever indicates that the virus might be excreted in faeces [84]. Ten days after admission, she underwent real-time PCR of a pharyngeal sample that was negative for SARS-CoV-2. A separate faecal sample was tested and found positive. The next 7 days 4 additional samples from the respiratory tract were tested and were all negative. This suggests that the gastrointestinal tract was the only documented source of the virus infection. In a recent Singaporean study, 50% of patients had SARS-CoV-2 detected in their stool samples, but detection did not correlate with the presence of digestive symptoms [85]. In another study, the duration of viral RNA detection after recovery was examined [86]. The median time from symptoms onset to first negative RT-PCR test from oropharyngeal swab was 9.5 days, but 16.7% of the patients tested positive for viral RNA from stool specimens for a median of 11 days. This observation indicates that there might be a potential faecal–oral transmission risk many days after symptoms resolution.
Regarding the liver-related complications of COVID-19, liver test abnormalities have been described in infected patients. In one study, it was demonstrated that total bilirubin, AST and ALT were elevated in 10%, 21% and 22% of patients, respectively [43]. Other case series have reported ALT abnormalities in 16–53% [40, 51, 87,88,89,90]. To date, cases of acute liver failure have not been reported. Liver dysfunction is mainly described in patients with severe disease upon presentation. However, it is difficult to discriminate the independent effect of the infection from other treatment modalities, such as antibiotics and antiviral drugs administered to these patients. Additionally, these abnormalities could be attributed to the infection itself, the induced sepsis or the concurrence of hypoxia. One patient underwent liver autopsy which revealed microvesicular steatosis, mild lobular and portal inflammation [91]. ACE2 receptors are located in hepatocytes and cholangiocytes; therefore, it was anticipated that the liver would also be involved. However, cholestatic abnormalities have rarely been described. Underlying pre-existing liver diseases could have contributed to liver enzyme abnormalities. The exact pattern of liver injury as well as its role in mortality needs to be further investigated. Finally, very recently a systemic review and meta-analyses were published in Lancet Gastroenterology and Hepatology in order to identify the prognosis and prevalence of digestive tract involvement and liver abnormalities in patients diagnosed with COVID-19. In total, 35 studies with 6686 patients were included in the analyses. The study demonstrated that gastrointestinal symptoms and hepatic toxicity are not uncommon among patients with COVID-19 disease [92].
Haematological manifestations
COVID-19 is a systemic infection with a significant impact on the haematopoietic system and homeostasis [93]. Lymphopenia may be considered as a cardinal laboratory finding, with prognostic potential. Approximately, 7–14 days from the onset of the initial symptoms, there is a surge in the clinical manifestations of the COVID-19 disease with a pronounced systemic increase in inflammatory mediators and cytokines, which may even be characterized as a “cytokine storm” [94]. In this context, significant lymphopenia becomes evident. Neutrophil/lymphocyte ratio and peak platelet/lymphocyte ratio may also have prognostic value in determining severe cases. Lymphocytes express the ACE2 receptor on their surface [81]; thus, SARS-CoV-2 may directly infect those cells, whereas the cytokine surge may promote lymphocyte apoptosis [95,96,97]. Substantial cytokine activation may be also associated with atrophy of lymphoid organs, including the spleen, and further impairs lymphocyte turnover [98]. Abnormalities in haematological parameters have been more prominent among severe versus non-severe cases (96.1% versus 80.4% for lymphocytopenia, 57.7% versus 31.6% for thrombocytopenia and 61.1% versus 28.1% for leukopenia). These results were consistent in four other descriptive studies that were conducted during the same period in China and included 41, 99, 138 and 201 confirmed cases with COVID-19, respectively [40, 42, 50, 87]. A meta-analysis of nine studies suggested that thrombocytopenia is significantly associated with the severity of the COVID-19 disease, with very high between-studies heterogeneity, though a more sizeable drop in platelet counts was noted especially in non-survivors [99]. During the disease course, longitudinal evaluation of lymphocyte count dynamics and inflammatory indices, including LDH, CRP and IL-6, may help to identify cases with dismal prognosis and prompt intervention in order to improve outcomes [93]. Biomarkers such as high serum procalcitonin, CRP and ferritin have also emerged as poor prognostic factors [41, 43, 100]. More recently, high cortisol levels at presentation may reflect disease severity and have been recognized as an adverse prognostic factor associated with poor survival among patients with severe COVID-19 [101].
Furthermore, blood hypercoagulability is common among hospitalized COVID-19 patients, especially among those with severe disease [41, 102, 103]. Elevated D-dimer levels are consistently reported, whereas their gradual increase during disease course is associated with clinical deterioration [40, 43, 50, 87, 104]. Other coagulation abnormalities such as PT and aPTT prolongation, increasing fibrin degradation products, with severe thrombocytopenia lead to life-threatening disseminated intravascular coagulation (DIC) which necessitates continuous vigilance and prompt intervention [41, 42, 52, 105,106,107,108]. Endothelial dysfunction and immune deregulation may be implicated in the underlying pathophysiology [109]. COVID-19 infected patients are at high risk of venous thromboembolism (VTE) (up to 10% for acutely ill hospitalized patients [110]). Comorbidities, along with the possibility of endothelial cell activation/damage due to the virus binding to ACE2 receptor, collectively increase the risk of VTE. Prompt pharmacological thromboprophylaxis with low molecular weight heparin is highly recommended [93, 111, 112].
Neurological manifestations
Although coronaviruses mainly cause respiratory symptoms, they have been reported to be involved in direct CNS infection as well as para-infectious complications [113]. Recent studies reported that over a third of infected patients developed a broad spectrum of neurological symptoms affecting central nervous system (CNS), peripheral nervous system (PNS) and skeletal muscles [114, 115]. In each case, it has to be noted that the challenge lies in discriminating between causal relationship and incidental comorbidity [116].
A large retrospective observational study from China showed that among 214 hospitalized patients with confirmed SARS-CoV-2 infection, 36.4% had neurological manifestations [114]. Most neurological symptoms occurred early during the first days after hospital admission. With regard to symptom category, 24.8% of infected patients presented symptoms from CNS, 8.9% from PNS and 10.7% developed skeletal muscle injury. The most common CNS symptoms were dizziness (16.8%) and headache (13.1%), and the most commonly reported symptoms involving PNS were taste impairment (5.6%) and anosmia (5.1%). Other less frequent symptoms included impaired consciousness (7.5%), acute cerebrovascular disease (2.8%), ataxia (0.5%), seizure (0.5%), vision impairment (1.4%) and nerve pain (2.7%). Notably among severely infected patients the prevalence of neurological manifestation was even higher, up to 45% compared to patients with less severe disease (30.2%). The correlation of disease severity with neurological symptoms was confirmed by another retrospective study from France, reporting a prevalence of 84% of neurological manifestations in 58 hospitalized patients with acute respiratory distress syndrome (ARDS) due to COVID-19 [115]. Of note, some of the reported symptoms such as inattention, disorientation and movement disorders persisted even after discharge.
Several mechanisms that may overlap have been proposed to explain the link between SARS-CoV-2 infection and nervous system injury [117]. Clinical manifestations of COVID-19 might be a consequence of the viral infection per se and/or the adverse insult of the hyperinflammatory status and dysregulated metabolic function, in combination with the multiple organ damage observed in patients after COVID-19 infection [113, 118]. Direct viral damage of nervous tissue might be possible in different ways. As with SARS and MERS viruses, SARS-COV-2 may enter the CNS through the hematogenous or retrograde neuronal route. Infection of olfactory neurons in the nose may enable the virus to enter the brain transneuronally and spread directly from the respiratory tract to the brain [118].
ACE2 receptors are also found in the nervous system and skeletal muscles [119, 120]. The expression and distribution of ACE2 in brain and endothelial cells may explain how SARS-CoV-2 may cause direct neurological symptoms and skeletal muscle damage. Direct viral damage of nervous tissue resembling in some ways herpes simplex encephalitis might be also possible, although there is no definite evidence of direct injury of CNS by SARS-CoV-2 virus. The excessive immune response which results in a hyperinflammatory status and cytokine storm may represent another alternative mechanism. Cytokines can directly pass through the blood–brain barrier causing considerable damage such as acute necrotizing encephalopathy [121]. An indirect injury related to host immune response effects after acute coronavirus infection could also be possible, explaining to some extent the occurrence of Guillain-Barré syndrome (GBS) cases, transverse myelitis or acute disseminated encephalomyelitis in patients with COVID-19 and other virus epidemics [113, 122, 123]. Neurological symptoms caused by systemic illness especially in severely ill patients could also justify neurological manifestations of COVID-19 infection [114]. Patients admitted to intensive care unit (ICU) often develop encephalopathy, myopathy, autonomic neuropathy and polyneuromyopathy related to critical illness [124].
Cerebrovascular disease represents another mechanism explaining neurological signs and symptoms in COVID-19 patients, although the rate of acute stroke admissions has been significantly reduced over the COVID-19 pandemic [125]. A large retrospective study from China reported a rate of 5.4% for both haemorrhagic and ischaemic strokes among critically ill patients [114]. A recent study reported a rate of 0.9% imaging proven ischaemic stroke among 3556 hospitalized patients [126], stressing that cerebrovascular events may have been underestimated in intubated and sedated patients with severe COVID-19. In a recent study based on data from the Global COVID-19 Stroke Registry, it was shown that patients with stroke and concurrent COVID-19 infection had a higher risk of severe disability (P < 0.001) and death (odds ratio 4.3, 95% CI 2.22–8.30) compared with patients without COVID-19 [127]. Severe SARS-CoV-2 infection is a hypercoagulable state and may predispose to both venous and arterial thromboembolic events. Systemic inflammatory response triggers autoimmune mechanisms, leading to dysregulation of the coagulation cascade as reflected by elevated D-dimers, prolonged prothrombin time, high fibrinogen levels, low anti-thrombin levels, thrombocytopenia and diffuse intravascular coagulation in severely ill patients with COVID-19 [83, 106, 128]. Imbalance between procoagulant and anticoagulant homeostatic mechanisms may result in endothelial damage, microvascular thrombosis and vessel occlusion. In addition, cardiac dysregulation and cardiac arrhythmias/dysrhythmias attributed to excessive inflammation and to respiratory failure, may lead to cardiac strain and myocardial injury/dysfunction facilitating cardioembolism. Among COVID-19 patients who suffered a stroke, the rate of cryptogenic and embolic strokes was higher, and events were more severe and affected younger patients [126, 129]. Finally, blood pressure alterations, hypotension or hypertension, may lead to impaired cerebral perfusion and cerebrovascular events.
Anosmia and ageusia
Although not highlighted in the initial cohort studies, olfactory (OD) and gustatory (GD) sense dysfunctions have been reported as common symptoms of COVID-19 from several centres worldwide. Prevalence of smell and taste disturbances varies considerably depending mainly on the assessment criteria and tools used and on the degree of sense dysfunction.
In a recent multicentre European study [130], it was shown that patients with mild-to-moderate forms of COVID19 present commonly with OD (anosmia or hyposmia) and GD (hypogeusia or ageusia), (86% and 88%, respectively), even without nasal symptoms. Women were more likely to be affected, and there was an early olfactory recovery rate of 44%, while symptoms could last even 14 days after the resolution of symptoms. Another study reported a high prevalence of OD (61%) in COVID-19 patients, with an early and severe occurrence and a high correlation with loss of taste. OD was still found more prevalent in women, but also in younger patients, while there was an association with shortness of breath [131]. A pilot quantitative study has also shown that a moderate olfactory dysfunction is present in approximately three-quarters of hospitalized COVID-19 patients using an objective smelling identification test [132]. A multicentre prospective study demonstrated an under-reporting of OD and GD manifestations in patients with more severe COVID19 disease neglecting their symptomatology. These findings confirm that OD and GD are not predictors of a milder disease but are also markedly present in COVID19 severe infection [133].
The American Academy of Otolaryngology—Head and Neck Surgery and the British Association of Otorhinolaryngology suggested that anosmia–hyposmia and hypogeusia–ageusia should be considered as “significant symptoms” even in the absence of other nasal manifestations (rhinorrhea or nasal congestion), which should be used as potential markers of otherwise asymptomatic carriers of COVID-19 infection (i.e. as a screening tool). In particular, the sudden onset of olfactory dysfunction could represent an early indicator of COVID-19 infection [134]. These findings make clear that clinicians should take into account self-reporting OD and GD symptomatology and incorporate in their assessment, the evaluation of the olfactory nerve function [135].
Despite the lack of a clear pathogenetic mechanism explaining OD and GD manifestations in COVID19 patients, it seems that there is a specific viral neuroinvasivity and neutropism via the olfactory nerves spreading rapidly to other brain structures such as the thalamus and the brainstem, but also possibly to the temporal lobe, the amygdala, insula, limbic lobe (psycho sensorial syndrome) [88]. Neurotropism may also occur via circulation and/or an upper transnasal route COVID-19 to reach the brain tissue, where COVID-19 spike protein binds angiotensin-converting enzyme 2 (ACE2) receptors [136]. Interestingly, the presence of ACE-2, in host olfactory and gustatory pathways, might provide a potential explanatory mechanism for the smell and taste disorders in COVID-19 patients. The expression level of ACE2 in different tissues and in particular neural cells might be also important in viral neurotropism differences between patients from different geographic regions. Thus, the differential ACE2 expression could give an explanation of the higher prevalence of OD and GD observed in European compared to Asian population [114]; however, more studies are needed to confirm such hypothesis.
Kidney involvement
In the kidney, ACE2 is present in several cells such as podocytes, mesangial cells, epithelium of the Bowman’s capsule, proximal cells brush border and collecting ducts [119]. The most frequent abnormality in patients with COVID-19 is mild-to-moderate proteinuria which is mediated via several mechanisms [137]. It has been reported that patients in the ICU have higher levels of IL-1β, IL-8, IFN-γ and TNF-α [40]. This suggests a potential role of cytokine release syndrome (CRS), also known as “cytokine storm” comparable with sepsis-associated AKI (SA-AKI), where the uncontrolled systemic inflammatory response leads to kidney injury [40]. Other studies have confirmed tropism to monocytes as well as lymphocytes, where the virus induces proinflammatory responses and cell death [138]. In addition, alterations in renal haemodynamics can induce further dysfunction [139]. Acute kidney injury (AKI) is infrequent in patients with mild-to-moderate disease (5%). In this patient subgroup, the abnormalities are mainly subclinical. A recent prospective study, which included 701 patients with moderate or severe disease, demonstrated that 43.9% presented with proteinuria and 26.7% with haematuria at hospital admission. Thirteen percentage revealed elevated levels of either serum creatinine (SCr), blood urea nitrogen (BUN) or both. AKI occurred in 5.1% of hospitalized patients. All these abnormalities conferred for higher death risk [140]. Another recent report showed that AKI was more common in critically ill patients. In 52 critically ill patients who were admitted to an intensive care unit (ICU) in Wuhan, AKI was the most common extra-pulmonary complication, occurring in 15 patients (29%). Eight patients (25%) required continuous renal replacement therapy, and 12 (80%) died with a median duration from admission to ICU until death of 7 days [51]. In another study previously described, a in silico analysis of publicly available data sets of single-cell RNA sequencing was performed. This analysis showed that RNA for angiotensin-converting enzyme 2 (ACE2), transmembrane serine protease 2 (TMPRSS2) and cathepsin L (CTSL) is enriched in several kidney cells. This enrichment may explain the relevant affinity that induces SARS-CoV-2 kidney injury [3]. Following that, tissue microdissection was applied on 6 kidneys biopsied to define SARS-CoV-2 viral load in exact kidney compartments. Three patients revealed detectable SARS-CoV-2 viral load in all compartments examined, mainly at the glomerular cells. These extremely interesting findings indicate that renal tropism is the obvious reason leading to kidney injury, even in the absence of severe disease. These data indicate that kidney abnormalities are common and are associated with worse clinical outcomes. Kidney autopsies of SARS-CoV patients have also demonstrated that the virus was present in tubular epithelial cells [141].
Skin manifestations
A number of case reports on skin complications observed in patients with COVID-19 have been published. Skin abnormalities are seen in up to 20% of COVID-19 patients in some series and are very heterogenous ranging from urticarial, vesicular, purpuric to papulosquamous lesions. It is, however, not clear currently whether these skin manifestations are caused directly by the virus invasion or secondary to host immune response or treatment administration. Purpuric eruptions, livedo reticularis or retiform purpura could be part of the manifestations of the vasculopathy associated with COVID-19 infection [142]. The nature of the association between COVID-19 and skin lesions and the systemic implications of their presence remains to be determined and requires active input and effort from dermatologists [143,144,145,146].
Endocrine abnormalities
Knocking down the host’s response to cortisol stress is a strategy employed by many viruses, including SARS-CoV to evade the host immune system. SARS-CoV expresses key amino acids that act as molecular mimics to the host adrenocorticotropic hormone (ACTH) directing antibodies to these ACTH residues implying a relative cortisol insufficiency. Data on serum cortisol levels in SARS-CoV-2 patients are scarce to date. A recent study among patients with no signs of adrenal insufficiency showed that high cortisol levels at presentation may reflect systemic disease severity and have associated with dismal survival among patients with severe COVID-19 [101]. Autopsy studies from SARS-CoV viral infection have demonstrated degeneration and necrosis of the adrenal gland, and the virus has been identified in the glands themselves pointing to the likelihood that cortisol dynamics are altered in SARS patients. The hypothalamic–pituitary–adrenal (HPA) axis might also be affected by SARS viruses on the ground of a reversible hypophysitis or direct hypothalamic damage. ACE2 is expressed on both hypothalamic and pituitary tissues explaining a possible viral tropism. A prospective study (ChiCTR20000301150) is currently evaluating serum cortisol and ACTH levels in COVID-19 patients [147,148,149].
Opthalmological complications
In animals, coronaviruses have been known to cause ocular manifestations including conjunctivitis, uveitis, retinitis and even optic neuritis [150]. In humans, the eye conjunctiva is considered to be a potential site for SARS-CoV-2 transmission [151], but currently there is no direct evidence to support that viral replication can cause injury and inflammation of the conjunctiva or other eye parts. Among 38 COVID-19 infected patients, in the Hubei province case report series, 12 had ocular manifestations (31.6%). These were more common among patients with more severe systemic disease presentation (respiratory mostly) and blood test abnormalities. They included conjunctival congestion, chemosis or epiphora [152]. A recent protocol used optical coherence tomography to evaluate the retina of patients with COVID-19 infection in 12 adults. Hyper-reflective lesions of the inner plexiform layers and the ganglion cells were seen in all patients, and cotton wool spots and microhemorrhages in the retinal arcade of 4 patients with no effect on visual acuity or pupillary reflexes [153]. Increasingly emerging data will allow better understanding of the nature and the mechanisms underlying the ocular manifestations associated with SARS-CoV-2.
Concluding remarks
COVID-19 probably represents the greatest pandemic event in modern human history. The disease presents with a broad spectrum of clinical signs and symptoms with involvement of vital organs such as the lungs, the heart, the gastrointestinal tract, the liver, the central nervous system, the blood and the kidneys. Commonly, multisystemic involvement is associated with severe disease and might predict worse clinical outcomes and increased mortality. The main mechanism described is the high binding affinity of the virus with the ACE2 receptors that are widely expressed in most human cells. The exact role of ACE2 receptors in COVID-19 pathophysiology is part of ongoing investigations. Furthermore, the role of the infection on dysregulation of ACE2 receptors expression, whether treatment with ARBs and ACEs modifies this expression and whether patients with comorbidities and chronic illnesses have higher expression of ACE2 receptors and are therefore more vulnerable to infection are also questions that need to be addressed in the near future. The SARS-CoV-2 virus enters the body through the respiratory tract and infects the epithelial cells of the trachea, bronchi, bronchioles and finally the lungs. It then infects the host, and infiltrating and circulating immune cells transfer the virus to other organs. Moreover, the blood-borne SARS virus infects other organs as well. Immunosuppressed patients, including the elderly and patients with chronic disease, experience more severe disease with increased mortality rates. The extent of immune cell damage, represented by the lymphocyte count, is considered a strong predictor of outcome and reflects the immune status of the patient. This multisystemic disease is associated with high mortality rates; mechanical ventilation, extracorporeal membrane oxygenation, antivirals and plasma infusion are currently being applied to reduce mortality, but none is a curative intervention. Although systematic treatments are currently at the forefront of clinical research, organ-specific treatment strategies should be also evaluated in order to optimize the management of patients with severe organ dysfunction. Several clinical trials are ongoing to evaluate the safety and effectiveness of both novel and pre-existing antiviral drugs, but the ability to vaccinate people will require unfortunately more time. The unpredictable trajectory of this unexpected pandemic requires careful surveillance, customized health strategies, control measures implementation, novel legal and bioethical framework, and specific medical guidelines to guide our decisions.
References
Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181–92.
de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523–34.
Puelles VG, Lutgehetmann M, Lindenmeyer MT et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med. 2020.
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA. 2020.
Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323:1061–9.
Goyal P, Choi JJ, Pinheiro LC et al. Clinical characteristics of Covid-19 in New York City. N Engl J Med. 2020;382(24):2372–2374.
Bhatraju PK, Ghassemieh BJ, Nichols M et al. Covid-19 in critically ill patients in the Seattle region—case series. N Engl J Med. 2020;382(21):2012–2022.
Wang Y, Lu X, Chen H et al. Clinical course and outcomes of 344 intensive care patients with COVID-19. Am J Respir Crit Care Med. 2020.
Wan Y, Shang J, Graham R, et al. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94:e00127-00120.
Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–4.
Channappanavar R, Zhao J, Perlman S. T cell-mediated immune response to respiratory coronaviruses. Immunol Res. 2014;59:118–28.
Huang SH. What we know so far (as of March 26, 2020) about COVID-19-an MRT point of view. J Med Imaging Radiat Sci. 2020;51(2):200–203.
Li H, Liu L, Zhang D, et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 2020;395:1517–20.
Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420–2.
Xu B, Fan CY, Wang AL et al. Suppressed T cell-mediated immunity in patients with COVID-19: a clinical retrospective study in Wuhan, China. J Infect. 2020;81(1):e51–e60.
Zhou G, Chen S, Chen Z. Advances in COVID-19: the virus, the pathogenesis, and evidence-based control and therapeutic strategies. Front Med. 2020;14(2):117–125.
Buja LM, Wolf D, Zhao B et al. Emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): report of three autopsies from Houston, Texas and review of autopsy findings from other United States Cities. Cardiovasc Pathol. 2020; (published online).
Barton LM, Duval EJ, Stroberg E et al. COVID-19 autopsies, Oklahoma, USA. Am J Clin Pathol. 2020;153(6):725–733.
McGonagle D, O’Donnell JS, Sharif K et al. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet. 2020.
Wolfel R, Corman VM, Guggemos W et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020.
Guan W, Liu J, Yu C. CT Findings of coronavirus disease (COVID-19) severe pneumonia. AJR Am J Roentgenol. 2020;214:W85–6.
Chen X, Liu S, Zhang C, et al. Dynamic chest CT evaluation in three cases of 2019 novel coronavirus pneumonia. Arch Iran Med. 2020;23:277–80.
Huang G, Gong T, Wang G et al. Timely diagnosis and treatment shortens the time to resolution of coronavirus disease (COVID-19) pneumonia and lowers the highest and last CT scores from sequential chest CT. AJR Am J Roentgenol. 2020; 1–7.
Zhao W, Zhong Z, Xie X, et al. CT scans of patients with 2019 novel coronavirus (COVID-19) pneumonia. Theranostics. 2020;10:4606–13.
Simpson S, Kay FU, Abbara S et al. Radiological society of north america expert consensus statement on reporting chest CT findings related to COVID-19. Endorsed by the Society of Thoracic Radiology, the American College of Radiology, and RSNA. J Thorac Imaging. 2020;35(4):219–227.
Bai HX, Hsieh B, Xiong Z et al. Performance of radiologists in differentiating COVID-19 from viral pneumonia on chest CT. Radiology. 2020;296(2):E46–E54.
Wang J, Xu Z, Wang J, et al. CT characteristics of patients infected with 2019 novel coronavirus: association with clinical type. Clin Radiol. 2020;75:408–14.
Han R, Huang L, Jiang H, Early clinical and CT manifestations of coronavirus disease, et al. (COVID-19) Pneumonia. AJR Am J Roentgenol. 2019;2020:1–6.
Ottestad W, Seim M, Maehlen JO. COVID-19 with silent hypoxemia. Tidsskr Nor Laegeforen. 2020; 140.
Gattinoni L, Coppola S, Cressoni M et al. COVID-19 does not lead to a “typical” acute respiratory distress syndrome. Am J Respir Crit Care Med. 2020;201(10):1299–1300.
Gattinoni L, Chiumello D, Caironi P et al. COVID-19 pneumonia: different respiratory treatments for different phenotypes? Intens Care Med. 2020;46(6):1099–1102.
Gattinoni L, Caironi P, Cressoni M, et al. Lung recruitment in patients with the acute respiratory distress syndrome. N Engl J Med. 2006;354:1775–86.
Pan C, Chen L, Lu C et al. Lung recruitability in SARS-CoV-2 associated acute respiratory distress syndrome: a single-center, observational study. Am J Respir Crit Care Med. 2020;201(10):1294–1297.
Caputo ND, Strayer RJ, Levitan R. Early self-proning in awake, non-intubated patients in the emergency department: a single ED’s experience during the COVID-19 pandemic. Acad Emerg Med. 2020;27:375–8.
Ragab D, Salah Eldin H, Taeimah M, et al. The COVID-19 cytokine storm; what we know so far. Front Immunol. 2020;11:1446.
Solinas C, Perra L, Aiello M et al. A critical evaluation of glucocorticoids in the management of severe COVID-19. Cytokine growth factor Rev. 2020;S135-6101(20)30161–1.
Antony SJ, Davis MA, Davis MG et al. Early use of tocilizumab in the prevention of adult respiratory failure in SARS-CoV-2 infections and the utilization of interleukin-6 levels in the management. J Med Virol. 2020. https://doi.org/10.1002/jmv.26288.
Titanji BK, Farley MM, Mehta A et al. Use of Baricitinib in Patients with Moderate and Severe COVID-19. Clin Infect Dis. 2020;ciaa879.
Horby P, Lim W, Emberson J, et al. Effect of dexamethasone in hospitalized patients with COVID-19: preliminary report. medRxiv. 2020. https://doi.org/10.1101/2020.06.22.20137273.
Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506.
Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62.
Wu C, Chen X, Cai Y et al. Risk Factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020
Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708–20.
Ruan Q, Yang K, Wang W, et al. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46:846–8.
Huang R, Zhu L, Xue L, et al. Clinical findings of patients with coronavirus disease 2019 in Jiangsu province, China: a retrospective, multi-center study. PLoS Negl Trop Dis. 2020;14:e0008280.
Wang W, Xu Y, Gao R et al. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA. 2020;323(18):1843–1844.
Guan WJ, Liang WH, Zhao Y et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: a nationwide analysis. Eur Respir J. 2020;55(5):2000547.
Zhang JJ, Dong X, Cao YY et al. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy. 2020;75(7):1730–1741.
Richardson S, Hirsch JS, Narasimhan M et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City Area. JAMA. 2020;323(20):2052–2059.
Wang D, Hu B, Hu C et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061–1069.
Yang X, Yu Y, Xu J, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8:475–81.
Guo T, Fan Y, Chen M et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5(7):1–8.
Chen T, Wu D, Chen H, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020;368:m1091.
Fu L, Wang B, Yuan T, et al. Clinical characteristics of coronavirus disease 2019 (COVID-19) in China: a systematic review and meta-analysis. J Infect. 2020;80:656–65.
Sanchis-Gomar F, Lavie C, Mehra M et al. Obesity and outcomes in COVID-19: when an epidemic and pandemic collide. Mayo Clinic Proc. 2020; (in press)
Gembardt F, Sterner-Kock A, Imboden H, et al. Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides. 2005;26:1270–7.
Hu H, Ma F, Wei X, Fang Y. Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. Eur Heart J. 2020;ehaa190.
Zeng JH, Liu YX, Yuan J et al. First case of COVID-19 complicated with fulminant myocarditis: a case report and insights. Infection. 2020;1–5.
Inciardi RM, Lupi L, Zaccone G et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;174:30–33.
Doyen D, Moceri P, Ducreux D, Dellamonica J. Myocarditis in a patient with COVID-19: a cause of raised troponin and ECG changes. Lancet. 2020;395:1516.
Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1-7 axis of the renin-angiotensin system in heart failure. Circ Res. 2016;118:1313–26.
Jiang F, Yang J, Zhang Y, et al. Angiotensin-converting enzyme 2 and angiotensin 1-7: novel therapeutic targets. Nat Rev Cardiol. 2014;11:413–26.
Alifano M, Alifano P, Forgez P, Iannelli A. Renin-angiotensin system at the heart of COVID-19 pandemic. Biochimie. 2020;174:30–3.
Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76:14–20.
South AM, Diz DI, Chappell MC. COVID-19, ACE2, and the cardiovascular consequences. Am J Physiol Heart Circ Physiol. 2020;318:H1084–90.
Qin C, Zhou L, Hu Z et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. 2020;ciaa248.
Bonow RO, Fonarow GC, O’Gara PT, Yancy CW. Association of coronavirus disease 2019 (COVID-19) with myocardial injury and mortality. JAMA Cardiol. 2020.
Libby P. The Heart in COVID19: primary target or secondary bystander? JACC Basic Transl Sci. 2020;5(5):537–542. https://doi.org/10.1016/j.jacbts.2020.04.001.
Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–8.
Han H, Xie L, Liu R et al. Analysis of heart injury laboratory parameters in 273 COVID-19 patients in one hospital in Wuhan, China. J Med Virol. 2020;92(7):819–823. https://doi.org/10.1002/jmv.25809
Musher DM, Abers MS, Corrales-Medina VF. Acute Infection and Myocardial Infarction. N Engl J Med. 2019;380:171–6.
Felker GM, Boehmer JP, Hruban RH, et al. Echocardiographic findings in fulminant and acute myocarditis. J Am Coll Cardiol. 2000;36:227–32.
Ammirati E, Cipriani M, Moro C, et al. Clinical presentation and outcome in a contemporary cohort of patients with acute myocarditis: multicenter Lombardy registry. Circulation. 2018;138:1088–99.
Gautret P, Lagier JC, Parola P et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents 2020;105949. https://doi.org/10.1016/j.ijantimicag.2020.105949.
Giudicessi JR, Noseworthy PA, Friedman PA, Ackerman MJ. Urgent Guidance for Navigating and Circumventing the QTc-Prolonging and Torsadogenic Potential of Possible Pharmacotherapies for Coronavirus Disease 19 (COVID-19). Mayo Clin Proc 2020;95(6):1213–1221.
Viner RM, Whittaker E. Kawasaki-like disease: emerging complication during the COVID-19 pandemic. Lancet 2020.
Verdoni L, Mazza A, Gervasoni A et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet 2020. https://doi.org/10.1016/S0140-6736(20)31103-X.
Liang W, Feng Z, Rao S, et al. Diarrhoea may be underestimated: a missing link in 2019 novel coronavirus. Gut. 2020;69:1141–3.
Ong J, Young BE, Ong S. COVID-19 in gastroenterology: a clinical perspective. Gut. 2020;69:1144–5.
Chen Y, Guo Y, Pan Y, Zhao ZJ. Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun 2020; 135–140.
Luo S, Zhang X, Xu H. Don’t overlook digestive symptoms in patients with 2019 novel coronavirus disease (COVID-19). Clin Gastroenterol Hepatol 2020.
Pan L, Mu M, Yang P, et al. Clinical characteristics of COVID-19 patients with digestive symptoms in Hubei, China: a descriptive, cross-sectional, multicenter study. Am J Gastroenterol. 2020;115:766–73.
Jin X, Lian JS, Hu JH, et al. Epidemiological, clinical and virological characteristics of 74 cases of coronavirus-infected disease 2019 (COVID-19) with gastrointestinal symptoms. Gut. 2020;69:1002–9.
Chen L, Lou J, Bai Y, Wang M. COVID-19 Disease With Positive Fecal and Negative Pharyngeal and Sputum Viral Tests. Am J Gastroenterol. 2020;115:790.
Young BE, Ong SWX, Kalimuddin S et al. Epidemiologic features and clinical course of patients infected with SARS-CoV-2 in Singapore. JAMA 2020;.23(15):1488–1494. https://doi.org/10.1001/jama.2020.3204.
Ling Y, Xu SB, Lin YX, et al. Persistence and clearance of viral RNA in 2019 novel coronavirus disease rehabilitation patients. Chin Med J (Engl). 2020;133:1039–43.
Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–13.
Shi H, Han X, Jiang N, et al. Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect Dis. 2020;20:425–34.
Zhang C, Shi L, Wang FS. Liver injury in COVID-19: management and challenges. Lancet Gastroenterol Hepatol. 2020;5:428–30.
Xu XW, Wu XX, Jiang XG, et al. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: retrospective case series. BMJ. 2020;368:m606.
Yao X, Ye F, Zhang M et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis 2020;ciaa237.
Mao R, Qiu Y, He JS et al. Manifestations and prognosis of gastrointestinal and liver involvement in patients with COVID-19: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2020;5(7):66–678.
Terpos E, Ntanasis-Stathopoulos I, Elalamy I et al. Hematological findings and complications of COVID-19. Am J Hematol 2020;95(7):834–847. https://doi.org/10.1002/ajh.25829.
Li T, Lu H, Zhang W. Clinical observation and management of COVID-19 patients. Emerg Microbes Infect. 2020;9:687–90.
Singh S, Sharma A, Arora SK. High producer haplotype (CAG) of -863C/A, -308G/A and -238G/A polymorphisms in the promoter region of TNF-alpha gene associate with enhanced apoptosis of lymphocytes in HIV-1 subtype C infected individuals from North India. PLoS ONE. 2014;9:e98020.
Liao YC, Liang WG, Chen FW, et al. IL-19 induces production of IL-6 and TNF-alpha and results in cell apoptosis through TNF-alpha. J Immunol. 2002;169:4288–97.
Aggarwal S, Gollapudi S, Gupta S. Increased TNF-alpha-induced apoptosis in lymphocytes from aged humans: changes in TNF-alpha receptor expression and activation of caspases. J Immunol. 1999;162:2154–61.
Chan JF, Zhang AJ, Yuan S et al. Simulation of the clinical and pathological manifestations of Coronavirus Disease 2019 (COVID-19) in golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin Infect Dis 2020;ciaa325. https://doi.org/10.1093/cid/ciaa325.
Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta. 2020;506:145–8.
Lippi G, Plebani M. Procalcitonin in patients with severe coronavirus disease 2019 (COVID-19): a meta-analysis. Clin Chim Acta. 2020;505:190–1.
Tan T, Khoo B, Mills EG et al. Association between high serum total cortisol concentrations and mortality from COVID-19. Lancet Diabetes Endocrinol. 2020;8(8):659–660. https://doi.org/10.1016/S2213-8587(20)30216-3.
Deng Y, Liu W, Liu K et al. Clinical characteristics of fatal and recovered cases of coronavirus disease 2019 (COVID-19) in Wuhan, China: a retrospective study. Chin Med J (Engl). 2020;133(11):1261–1267.
Levi M, Thachil J, Iba T, Levy JH. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020;7(6):e438–e440. https://doi.org/10.1016/S2352-3026(20)30145-9.
Snijders D, Schoorl M, Schoorl M, et al. D-dimer levels in assessing severity and clinical outcome in patients with community-acquired pneumonia. A secondary analysis of a randomised clinical trial. Eur J Intern Med. 2012;23:436–41.
Shi S, Qin M, Shen B et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020.
Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844–7.
Bloch EM, Shoham S, Casadevall A et al. Deployment of convalescent plasma for the prevention and treatment of COVID-19. J Clin Invest 2020;130(6):2757–2765.
Lippi G, Favaloro EJ. D-dimer is associated with severity of coronavirus disease 2019: a pooled analysis. Thromb Haemost 2020;120(5):87–878. https://doi.org/10.1055/s-0040-1709650.
Lillicrap D. Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J Thromb Haemost. 2020;18:786–7.
Kahn SR, Lim W, Dunn AS, et al. Prevention of VTE in nonsurgical patients: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e195S–226S.
Tang N, Bai H, Chen X et al. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18(5):1094–1099.
Thachil J, Tang N, Gando S et al. ISTH interim guidance on recognition and management of coagulopathy in COVID‐19. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14810.
Needham EJ, Chou SH, Coles AJ, Menon DK. Neurological implications of COVID-19 infections. Neurocrit Care. 2020;32:667–671.
Mao L, Jin H, Wang M et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020;77(6):1–9. https://doi.org/10.1001/jamaneurol.2020.1127.
Helms J, Kremer S, Merdji H et al. Neurologic features in severe SARS-CoV-2 infection. N Engl J Med 2020;382:2268–2270. https://doi.org/10.1056/NEJMc2008597.
Tsivgoulis G, Palaiodimou L, Katsanos AH, et al. Neurological manifestations and implications of COVID-19 pandemic. Ther Adv Neurol Disord. 2020;13:1756286420932036.
Zubair AS, McAlpine LS, Gardin T et al. Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: A Review. JAMA Neurol 2020.
Nath A. Neurologic complications of coronavirus infections. Neurology. 2020;94:809–10.
Hamming I, Timens W, Bulthuis ML, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7.
Cabello-Verrugio C, Morales MG, Rivera JC, et al. Renin-angiotensin system: an old player with novel functions in skeletal muscle. Med Res Rev. 2015;35:437–63.
Poyiadji N, Shahin G, Noujaim D et al. COVID-19-associated acute hemorrhagic necrotizing encephalopathy: CT and MRI features. Radiology 2020; 201187.
Toscano G, Palmerini F, Ravaglia S et al. Guillain-Barré syndrome associated with SARS-CoV-2. New Engl J Med. 2020; NEJMc2009191.
Zhao H, Shen D, Zhou H, et al. Guillain-Barré syndrome associated with SARS-CoV-2 infection: causality or coincidence? Lancet Neurol. 2020;19:383–4.
Vanhorebeek I, Latronico N, Van den Berghe G. ICU-acquired weakness. Intensive Care Med. 2020;46:637–53.
Kansagra AP, Goyal MS, Hamilton S, Albers GW. Collateral effect of COVID-19 on stroke evaluation in the United States. New Engl J Med. 2020.
Yaghi S, Ishida K, Torres J et al. SARS2-CoV-2 and stroke in a new york healthcare system. Stroke 2020; Strokeaha120030335.
Ntaios G, Michel P, Georgiopoulos G, et al. Characteristics and outcomes in patients with COVID-19 and acute ischemic stroke. The global COVID-19 stroke registry. Stroke. 2020. https://doi.org/10.1161/STROKEAHA.120.031208.
Bikdeli B, Madhavan MV, Jimenez D et al. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up. J Am Coll Cardiol. 2020.
Oxley TJ, Mocco J, Majidi S, et al. Large-vessel stroke as a presenting feature of COVID-19 in the young. N Engl J Med. 2020;382:e60.
Lechien JR, Chiesa-Estomba CM, De Siati DR et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study. Eur Arch Otorhinolaryngol 2020; 1–11.
Speth MM, Singer-Cornelius T, Obere M et al. Olfactory Dysfunction and Sinonasal Symptomatology in COVID-19: Prevalence, Severity, Timing, and Associated Characteristics. Otolaryngol Head Neck Surg. 2020; 194599820929185.
Tsivgoulis G, Fragkou PC, Delides A et al. Quantitative evaluation of olfactory dysfunction in hospitalized patients with Coronavirus [2] (COVID-19). J Neurol 2020; 1-3.
Vaira LA, Hopkins C, Salzano G et al. Olfactory and gustatory function impairment in COVID-19 patients: Italian objective multicenter-study. Head Neck 2020.
Lao WP, Imam SA, Nguyen SA. Anosmia, hyposmia, and dysgeusia as indicators for positive SARS-CoV-2 infection. World journal of otorhinolaryngology - head and neck surgery. 2020. https://doi.org/10.1016/j.wjorl.2020.1004.1001.
Giacomelli A, Pezzati L, Conti F et al. Self-reported olfactory and taste disorders in SARS-CoV-2 patients: a cross-sectional study. Clin Infect Dis 2020.
Baig AM, Khaleeq A, Ali U, Syeda H. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. 2020;11:995–8.
Cheng Y, Luo R, Wang K, et al. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020;97:829–38.
Gu J, Gong E, Zhang B, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202:415–24.
Rodriguez-Romo R, Benitez K, Barrera-Chimal J, et al. AT1 receptor antagonism before ischemia prevents the transition of acute kidney injury to chronic kidney disease. Kidney Int. 2016;89:363–73.
Boulware DR, Pullen MF, Bangdiwala AS et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for COVID-19. N Engl J Med 2020.
Ding Y, He L, Zhang Q, et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol. 2004;203:622–30.
Guarneri C, Rullo EV, Pavone P et al. Silent COVID-19: what your skin can reveal. Lancet Infect Dis. 2020. https://doi.org/10.1016/S1473-3099(20)30402-3.
Madigan LM, Micheletti RG, Shinkai K. How dermatologists can learn and contribute at the leading edge of the COVID-19 global pandemic. JAMA Dermatol. 2020. https://doi.org/10.1001/jamadermatol.2020.1438.
Sanchez A, Sohier P, Benghanem S et al. Digitate papulosquamous eruption associated with severe acute respiratory syndrome coronavirus 2 infection. JAMA Dermatol. 2020. https://doi.org/10.1001/jamadermatol.2020.1704.
Diaz-Guimaraens B, Dominguez-Santas M, Suarez-Valle A et al. Petechial skin rash associated with severe acute respiratory syndrome coronavirus 2 infection. JAMA Dermatol. 2020. https://doi.org/10.1001/jamadermatol.2020.1741.
Recalcati S. Cutaneous manifestations in COVID-19: a first perspective. J Eur Acad Dermatol Venereol. 2020;;34(5):e21–e213. https://doi.org/10.1111/jdv.16387.
Wheatland R. Molecular mimicry of ACTH in SARS—implications for corticosteroid treatment and prophylaxis. Med Hypotheses. 2004;63:855–62.
Leow MK, Kwek DS, Ng AW, et al. Hypocortisolism in survivors of severe acute respiratory syndrome (SARS). Clin Endocrinol (Oxf). 2005;63:197–202.
Pal R. COVID-19, hypothalamo-pituitary-adrenal axis and clinical implications. Endocrine 2020;28:1–2. https://doi.org/10.1007/s12020-020-02325-1.
Seah I, Agrawal R. Can the coronavirus disease 2019 (COVID-19) affect the eyes? A review of coronaviruses and ocular implications in humans and animals. Ocul Immunol Inflamm. 2020;28:391–5.
Peiris JSM, Yuen KY, Osterhaus ADME, Stöhr K. The severe acute respiratory syndrome. N Engl J Med. 2003;349:2431–41.
Wu P, Duan F, Luo C, et al. Characteristics of Ocular Findings of Patients With Coronavirus Disease 2019 (COVID-19) in Hubei Province, China. JAMA Ophthalmol. 2020;138:575–8.
Marinho PM, Marcos AA, Romano AC et al. Retinal findings in patients with COVID-19. Lancet 2020; (online first).
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no relevant conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gavriatopoulou, M., Korompoki, E., Fotiou, D. et al. Organ-specific manifestations of COVID-19 infection. Clin Exp Med 20, 493–506 (2020). https://doi.org/10.1007/s10238-020-00648-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10238-020-00648-x