
Abstract
Lymph node metastasis (LNM) is a usual event in papillary thyroid carcinoma (PTC) patients, which usually leads to poor prognosis. However, the molecular mechanisms of LNM remain unclear. Thus, we aimed to screen the possible key genes in the progression of LNM in PTC patients and further validate their roles. The study involved two phases: a discovery phase and a validation one. In the former phase, the candidate genes were screened by using bioinformatics methods. In the latter one, the genes were firstly assessed in a cohort from the cancer genome atlas (TCGA) to evaluate the associations of their expressions with clinical features and the prognostic values, and then, they were assessed at protein levels by using an immunohistochemical assay. Consequently, IGHBP3 was selected as the candidate gene, which might be enriched in several metabolism-related pathways and cancer progression-related pathways. High expressions of IGHBP3 have an association with gender, advanced clinical stages, high T stages, and the presence of LNM. Survival analysis indicated that IGHBP3 may affect the prognosis of PTC patients. The use of a tissue chip confirmed the view that IGHBP3 might play a crucial role in the LNM of PTC. In conclusion, IGHBP3 might be involved in the development of LNM in PTC patients. IGHBP3 over-expression might be a novel indicator and a potential target for PTC therapy.
Similar content being viewed by others

Background
Thyroid carcinoma is the most common endocrine malignant disorder in the world, and its incidence has increased in recent decades. Thyroid cancer can mainly be histologically classified into three types, namely, follicular thyroid cancer, papillary thyroid cancer (PTC), and anaplastic thyroid cancer [1]. The former two are well-differentiated, while the latter one is undifferentiated. Thus, the substantial characteristics of these pathological types are different. Of these types, PTC is the most common one, occupying about 80% of all thyroid cancers [2].
As a well-differentiated type of thyroid neoplasm, PTC often shows a favorable prognosis, especially in patients with young ages [3]. Nevertheless, a proportion of PTC patients still have unfavorable prognosis. Reports showed that 50% of PTC cases are diagnosed with cervical lymph node metastasis (LNM), suggesting that high frequency of LNM is a usual event in PTC patients, which may be the leading cause of poor prognosis for these patients [4, 5]. However, the molecular mechanisms underlying LNM remain little understood.
Reports have implicated the roles of a number of genes in the LNM of PTC. For example, over-expressions of IDH2 [6], VEGF, and Survivin [7] may be significantly correlated with the neck LNM in PTC patients. NECTIN4 is also over-expressed in PTC tissues, which may regulate the expression of epithelial-mesenchymal transition-related genes through activation of the PI3K/AKT pathway, and thus, up-regulated NECTIN4 is connected with LNM [8]. Similarly, TC-1 protein expression showed significant associations with LNM in PTC patients [9]. Nevertheless, these studies only focused on any single gene or pathway, neglecting that cancer development involves multiple gene variations, or dysregulation and activation of multiple signaling pathways [10].
To comprehensively learn the genes that might be critical for cancer progression, mining the high throughput data is a good strategy. With the development of microarray and sequencing, much data has been generated and stored in the public databases. Excavation of the disorganized data can help us find genes that are important in the development of PTC, and help us explore their roles in different biological processes.
To address this issue, we aimed to screen possible candidate genes and assess their possible roles in the development of LMN in PTC patients. The present study included two phases: a discovery phase and a validation one. In the discovery phase, we planned to screen the possible key genes that play critical roles in the process of PTC LNM by using bioinformatics approaches. In the validation phase, the roles of the screened genes were further assessed in a PTC cohort from the cancer genome atlas (TCGA) [11] to show their relations with clinical features as well as to evaluate their prognostic values in PTC patients. Then, the protein expressions of these genes were further assessed by an immunohistochemical (IHC) assay.
Materials and methods
Data screening
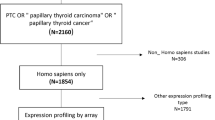
A dataset, GSE60542, was retrieved from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The type of the study was ‘Expression profiling by array’, which was confined to human species and conducted on the Affymetrix Human Genome U133 Plus 2.0 Array. The dataset was deposited by Tarabichi et al. [12], which contained 19 PTC samples from patients with LNM, and 14 samples from those without LNM.
The dataset was analyzed by using GEO2R that is a web tool based on the limma packages of R software [13]. The samples were divided into two groups according to the N stage, and a comparison of the two groups was carried out. The results were downloaded and analyzed, in which the genes that met the following cut–off criteria were screened out as the differentially expressed genes (DEGs): p < 0.05 and |log fold–change| > 1.0.
Assessment of the DEGs in UALCAN and Oncomine database
To screen the key genes that might have an influence on the prognosis of thyroid cancer, a cohort from the TCGA database was used for validation by using UALCAN tool [14]. The UALCAN is a convenient web-portal that helps researchers to carry out multiple analyses of TCGA gene expression data, which contains Level 3 RNA-seq data and relevant clinical information from a total of 31 carcinoma types. The prognostic value of each hub gene was individually evaluated. The genes whose expression levels affected the prognosis of thyroid cancer were regarded as the key genes.
Besides the UALCAN tool, the mRNA expression levels of the genes were also assessed in Oncomine tool [15]. The Oncomine is also a platform that provides researchers with a powerful analytical function for computing gene expression characteristics, clusters and gene set modules.
The gene expression level of the candidate genes was evaluated in the Oncomine and UALCAN, respectively. The mRNA levels of each gene in PTC and normal samples were compared.
Data mining in TCGA database
In order to explore the roles of the key genes in PTC cases, we conducted data excavation in the TCGA database. Standardized gene expression levels of the key genes in a TCGA cohort and relevant clinical data of PTC patients were obtained from TCGA and cBio Cancer Genomics Portal [16]. Samples with gene expression data, clinical characteristics data and prognosis data were achieved. Genes were divided as “high” and “low” groups in the light of the median expression levels, respectively.
Functional annotation and gene set enrichment analysis (GSEA)
To learn the possible biological functions of the key genes, they were submitted to Genecards tool [17] for evaluation. The biological process part of GO (gene ontology) and KEGG pathway enrichment analysis were concerned.
To explore the possible molecular mechanisms underlying the key genes, GSEA was also conducted to identify the significant common pathways the genes enriched in. TCGA data can be ordered in a ranked list according to the varied expression between the phenotypes. Each of the key gene level was divided, respectively, as high and low categories according to the median expression value to annotate phenotype. Gene set c2.cp.kegg.v6.2.symbols.gmt (Version 6.2) was chosen as the reference set. False discovery rate (FDR) < 0.05 was considered as the cut-off standard.
PTC specimens
PTC tissue microarray (HThy-Pap120CS-01) was purchased from Shanghai Outdo Biotech Co., Ltd. A total of 58 PTC samples and 58 paired adjacent non-cancer ones were included. Moreover, four cases of normal thyroid tissues were also included. All patients were pathologically diagnosed as PTC. No other treatment was performed before the operation. All samples were obtained after informed consent from the patients.
IHC staining and evaluation
The protein expressions of the key genes were detected by utilizing the two-step method of IHC as previously described in our study [18]. In brief, the slides were deparaffinized, rehydrated and treated with 3% hydrogen peroxided for 20 min to inhibit endogenous peroxidase. The slices were washed with distilled water and saturated in phosphate buffer (PBS) for 5 min, and then incubated overnight with goat anti-polyclonal antibody (primary antibody; ABCAM) at 4 °C. DAB solution was used to develop and hematoxylin was re-stained. IHC staining was performed according to the manufacturer’s instructions.
The results of IHC staining were identified by a comprehensive scoring method. Two pathologists independently evaluated the results without knowing the clinical parameters of the cases. The dyeing intensity of protein ranged from 0 to 3 points, in which 0 was negative, 1 was weak, 2 was medium, and 3 was strong. The percentage score of positive cells was 0–4, including 1 (0–25%), 2 (26–50%), 3 (51–75%), and 4 (76–100%). Then, multiply the proportion and intensity scores to get a total score, ranging from 0 to 12.
Statistical analysis
For continuous variables, Variance analysis (ANOVA), t test or Wilcoxon rank sum test were used to analyze the differences between the two groups according to the concrete types of the data. Chi-square test was used to distinguish the incidence of different groups. Kaplan–Meier method was used to calculate the overall survival curve, and Log-rank test was used to determine the difference in survival rate. COX multivariate regression analysis was carried out to assess the effects of the confounding factors on survival. These analyses were performed by utilizing MedCalc software (15.2.2; Mariakerke, Belgium). p < 0.05 is considered to have statistical significance.
Results
Key genes screening from microarray-based datasets
The dataset GSE60542 comprises 19 PTC samples from patients with LNM, and 14 PTC samples from those without LNM. The comparison of the two groups generated 5 up-regulated and 21 down-regulated DEGs (Table 1, and Fig. 1A).

A Heatmap of the DEGs screened from GSE60542. The horizontal axis above stands for the name of sample; the left vertical axis stands for the names of DEGs. Red stands for up-regulated genes, while green stands for down-regulated genes. B Survival curves of the representative DEGs evaluated in the UALCAN tool. a Down-regulated genes. b Up-regulated genes
After we had submitted these DEGs to the UALCAN tool for evaluation, the data showed that only the expression of one gene, IGFBP3, may have an influence on the overall survival time of thyroid carcinoma patients (Fig. 1B). Thus,IGFBP3 was selected for further analysis.
Expression of IGFBP3 between thyroid cancer and normal tissues
To assess the expression levels of IGFBP3 mRNA between thyroid cancer and normal thyroid tissues, the data in the Oncomine and TCGA database were analyzed.
As shown in Fig. 2A, IGFBP3 mRNA was significantly over-expressed in thyroid cancer tissues relative to normal controls.

A Expression of IGHBP3 mRNA in the thyroid cancer and the controls, respectively. a Oncomine database, p < 0.05 cancer vs control; b UALCAN database, p < 0.05 cancer vs control. B The survival curves in PTC patients with high and low expression of IGHBP3. a Disease-free survival; p > 0.05; b Overall survival; p < 0.05. C GSEA showed that high IGHBP3 expression was positively correlated with several metabolism-related and cancer-related pathways
Relationship of Clinicopathological factors with IGFBP3 mRNA expression
The mRNA expression data of IGFBP3 in a PTC cohort and relevant clinical data were obtained from TCGA database. Table 2 lists the characteristics of the involved PTC cases.
The expression levels of IGFBP3 were separated as high and low groups on the basis of their median level. As shown in Table 2, the relationship between IGFBP3 mRNA level and clinicopathological characteristics was explored. The results revealed that IGFBP3 high expression may have an association with male patients, advanced clinical stages, high T stages, and LNM, respectively. No associations were observed in the comparisons regarding age, race, histological type, and distant metastasis.
Association of IGFBP3 mRNA expression with the prognosis of PTC
The prognostic value of IGFBP3 in PTC was evaluated. As shown in Fig. 2B, the log-rank test indicated that PTC cases with high IGFBP3 expression had a shorter overall survival time than those of cases with low expression (p < 0.05). Nevertheless, IGFBP3 seemed to exert little influence on the disease-free survival in PTC patients (p > 0.05). However, multivariate cox regression analysis failed to suggest IGFBP3 expression as an independent prognostic factor for PTC (data not shown).
GO and pathway enrichment analysis of IGFBP3
To comprehensively explore the functions of IGFBP3, the GO and KEGG pathway enrichment analysis were conducted.
Table 3 listed the top 5 GO terms and pathways, respectively. GO analysis suggested that IGFBP3 has a relationship with regulation of cell growth, osteoblast differentiation, negative regulation of protein phosphorylation, protein phosphorylation, and apoptotic process.
Pathway analysis suggested that IGFBP3 might have a relationship with gene expression, TP53 regulates transcription of cell death genes, glucose/energy metabolism, cellular senescence, and DNA Damage Response.
To know the potential roles of IGFBP3 more specifically in the development of thyroid cancer, GSEA was applied to identify the possible pathways it enriched in. The results showed that high IGFBP3 expression was mainly associated with two categories of pathways: metabolism-related and cancer progression-related signaling pathways. As shown in Fig. 2c, the former mainly included arachidonic acid metabolism, alpha linolenic acid metabolism, ether lipid metabolism, galactose metabolism, glycerophospholipid metabolism, and linoleic acid metabolism; the latter mainly contained VEGF signaling pathway, P53 signaling pathway, Notch signaling pathway, Gnrh signaling pathway, Fc Epsilon Ri signaling pathway, and Cell Cycle.
Protein expression of IGFBP3 determined by IHC
The results of IHC showed that specific staining was observed in the cytoplasm of the cancer and normal cells, respectively (Fig. 3A-b). The data also confirmed that the scores of IGFBP3 in PTC tissues was higher than those in para-carcinoma ones (p < 0.05), as shown in Fig. 3B-a.

A IGHBP3 expression in PTC tissues and adjacent normal tissues. a IGHBP3 protein expressions in a tissue chip (paired PTC tissues and adjacent normal tissues) were measured with IHC (× 10); b Representative examples of IGHBP3 expression in PTC tissues and adjacent normal tissues (× 200); c stands for cancer, while p stands for para-cancer. B The results from the tissue chip. a The IGHBP3 expression was higher in PTC tissues than that in the normal tissues (p < 0.05). b–d High IGHBP3 expression was related with advanced age (b), advanced clinical stage (c), and the presence of LNM (d), respectively (p < 0.05 for each comparison)
There were a total of 58 PTC samples on the slice (Fig. 3A-a). The characteristics of these cases were presented in Table 4, of which no cases presented distant metastasis. Moreover, the survival information was not available in the chip. Confounding factors such as clinical stages, LNM, and age can be addressed.
Interestingly, the data indicated that higher expression score of IGFBP3 was correlated with advanced clinical stages, LNM, and advanced ages, respectively (p < 0.05) (Fig. 3B).
Discussion
To find the key genes in PTC LNM, we conducted a research containing a discovery phase and a validation one. In the former phase, we screened several genes that might be involved in the LNM of PTC through bioinformatics analyses, and found that IGFBP3 may be a key candidate gene and its over-expression might have an association with poor prognosis of PTC patients. In the validation phase, we firstly assessed the expression of IGFBP3 in a PTC cohort from TCGA database, and found that high IGFBP3 expression may have a relationship with sex, advanced clinical stages, high T stages, and LNM, as well as shorter overall survival time in patients. IGFBP3 was predicted to be enriched in many pathways regarding metabolisms and cell signaling pathways. Then, IGFBP3 protein expression was tested by IHC in a tissue microarray containing PTC samples. The data confirmed that IGFBP3 was up-regulated in PTC tissues relative to the para-carcinoma tissues. Higher expression score of IGFBP3 was associated with advanced clinical stages, LNM, and advanced ages, respectively.
IGFBP‐3, Insulin‐like growth factor binding protein‐3, is a protein of high affinity binding protein family, which regulates the function of insulin-like growth factor. It can be found in various organs and tissues and plays multiple roles in various biological processes, such as cell proliferation, senescence, apoptosis, and epithelial-mesenchymal transition [19]. Thus, IGFBP‐3 has been implicated to play a role in tumor development. Evidence indicates that IGFBP‐3 can play different roles in different carcinomas. For instance, over-expression of IGFBP‐3 has an association with esophageal cancer promotion and predicts poor prognosis [20]. Likewise, high expression of IGFBP‐3 in glioma may be associated with tumor histology and poor prognosis [21]. Nevertheless, IGFBP‐3 acts as a tumor suppressor in a number of cancers. In lung cancer cells, IGFBP3 over-expression might induce cancer cell apoptosis and increase sensitivity of cells to cisplatin [22]. In liver cancer cells, IGFBP3 expression was lower relative to the normal cells. IGFBP3 upregulation may lead to cell apoptosis and reduced colony formation [23]. Hence, IGFBP3 presents its multifunctional roles in various cancers. For PTC, both bioinformatics analysis and validation experiments showed that IGFBP3 was over-expressed in cancer tissues compared to the normal tissues, suggesting that IGFBP3 acts as an oncogene in PTC development. Moreover, the data also indicate that IGFBP3 expression might exert any influence on the overall survival time of PTC patients.
Evidence showed that IGFBP3 may exert its biological effects, in a sex-specific manner, on diseases such as obesity [24] and osteoporosis [25]. The results of the present study presented that the rate of high IGFBP3 in men (60.98%, 75/123) was markedly higher than that in women (43.50%,144/331), indicating that IGFBP3 might also play a certain role in the PTC development in a sex-specific manner. Reports showed that estrogen may affect the bioactivity of IGF1 and the IGFBP3 expression levels [26]; and both estrogen and androgen treatment can obviously regulate the expressions of IGF pathway genes, including IGFBP3 [27]. This may help speculate the reasons for the unbalanced expression of IGFBP3 in PTC tissues between male and female cases.
In the discovery phase, IGFBP3 has been selected as a candidate gene that may be associated with LNM of PTC. In the validation phase, the data based on a TCGA cohort indicated that high IGFBP3 mRNA expression may have a correlation with advanced clinical stages, high T stages, and the presence of LNM. Then, the data from a tissue chip confirmed that high IGFBP3 protein expression may be associated with LNM. The results steadily confirmed a critical role of IGFBP3 in the process of LNM in PTC patients. However, little about the molecular mechanisms has been clearly shown to date. A report showed that IGFBP3 may promote LNM in oral cancer through activation of ERK signaling [28]. However, few studies regarding thyroid cancer on this issue could be searched. Hence, future lab experiments are needed to explore the mechanisms of IGFBP3 in the process of LNM.
To comprehensively learn the possible functions of IGFBP3 in PTC, we conducted analyses by using the Genecards and GSEA tool, respectively. Both GO and KEGG enrichment analysis suggested that IGFBP3 may be enriched in a variety of pathways concerning multiple biological processes. However, GSEA analysis suggested that IGFBP3 might be mainly enriched in several cancer progression-related and metabolism-related pathways. Reports have shown that VEGF signaling_pathway [29], P53 signaling pathway [30], Notch signaling pathway [31], and Gnrh signaling pathway [32] may be involved in cancer development. As for the metabolism-related pathways, evidence has shown that they may also be related with cancer aggression. For instance, alpha linolenic acid metabolism was associated with aggressive prostate carcinoma [33]. Arachidonic acid metabolism might be involved in hepatocellular cancer progression [34]. Therefore, in future investigations, these pathways resulted from GSEA analysis need to be considered to explore the mechanisms underlying PTC progression.
Several limitations might be addressed. First, in the key genes screening process, ‘P values’ but not ‘adjusted P values’ had been used for screening the DEGs. In this case, if the ‘adjusted P values’ was considered, few genes met the selection criteria. Thus, we had to relax the inclusion criteria and used ‘P values’ instead. This less rigorous operation might lower the credibility of the results. Nevertheless, subsequent validation processes may to some extent make the results convincible. Second, in the validation process, only data in the TCGA database and a tissue chip were used. Future experiments using cell lines or animal models are needed to further investigate the molecular mechanisms of IGFBP3 in PTC development. Third, an IHC assay against paraffin tissue sections was used to validate the protein expression of IGFBP3 in PTC tissues. More comprehensive approaches, such as an agnostic quantitative proteomics approach against fresh frozen tissue sections, may help further identify suitable protein candidates [35]. However, a relevant commercial tissue chip containing fresh frozen tissue sections could not be obtained from the company. Therefore, this approach might be utilized in future studies when the fresh frozen tissue sections are available.
Conclusion
In summary, the results of this study suggest that over-expression of IGFBP3 might be associated with LNM in PTC patients. In addition, it may affect the prognosis of PTC and may become a potential target for cancer treatment.
Abbreviations
- LNM:
-
Lymph node metastasis
- PTC:
-
Papillary thyroid carcinoma
- TCGA:
-
The cancer genome atlas
- IHC:
-
Immunohistochemical
- GEO:
-
Gene Expression Omnibus
- DEGs:
-
Differentially expressed genes
- GSEA:
-
Gene set enrichment analysis
- GO:
-
Gene ontology
- FDR:
-
False discovery rate
- PBS:
-
Phosphate buffer
- ANOVA:
-
Variance analysis
- IGFBP‐3:
-
Insulin‐like growth factor binding protein‐3
References
Yakushina VD, Lerner LV, Lavrov AV. Gene fusions in thyroid cancer. Thyroid. 2018;28(2):158–67. https://doi.org/10.1089/thy.2017.0318.
Wang P, Li W, Zhai B, et al. Integrating high-throughput microRNA and mRNA expression data to identify risk mRNA signature for pancreatic cancer prognosis. J Cell Biochem. 2019. https://doi.org/10.1002/jcb.29576.
Zhao Z, Zhang KN, Chai RC, et al. ADAMTSL4, a secreted glycoprotein, is a novel immune-related biomarker for primary glioblastoma multiforme. Dis Mark. 2019;2019:1802620. https://doi.org/10.1155/2019/1802620.
Ge MH, Jiang LH, Wen QL, et al. Preliminary screening and analysis of metastasis-related lncRNA and co-expressed papillary thyroid carcinoma mRNA. Oncol Lett. 2018;16(3):3715–25. https://doi.org/10.3892/ol.2018.9080.
Choi JE, Bae JS, Lim DJ, Jung SL, Jung CK. atypical histiocytoid cells and multinucleated giant cells in fine-needle aspiration cytology of the thyroid predict lymph node metastasis of papillary thyroid carcinoma. Cancers (Basel). 2019. https://doi.org/10.3390/cancers11060816.
Zhang J, Hu L, Wang H, et al. Functional analysis and clinical significance of the isocitrate dehydrogenase 2 gene in papillary thyroid carcinoma. Cancer Manag Res. 2019;11:3765–77. https://doi.org/10.2147/CMAR.S194920.
Zhang HY, Meng X, Du ZX, et al. Significance of survivin, caspase-3, and VEGF expression in thyroid carcinoma. Clin Exp Med. 2009;9(3):207–13. https://doi.org/10.1007/s10238-009-0031-7.
Hao RT, Zheng C, Wu CY, et al. NECTIN4 promotes papillary thyroid cancer cell proliferation, migration, and invasion and triggers EMT by activating AKT. Cancer Manag Res. 2019;11:2565–78. https://doi.org/10.2147/CMAR.S190332.
Zhang CL, Geng CH, Jian JJ, He M, Zhao HW, Li HY. Correlations of ultrasound and pathological features of thyroid carcinoma with TC-1 mRNA and protein expression. Eur Rev Med Pharmacol Sci. 2019;23(8):3440–6. https://doi.org/10.26355/eurrev_201904_17708.
Cheng SP, Chen MJ, Chien MN, Lin CH, Lee JJ, Liu CL. Overexpression of teneurin transmembrane protein 1 is a potential marker of disease progression in papillary thyroid carcinoma. Clin Exp Med. 2017;17(4):555–64. https://doi.org/10.1007/s10238-016-0445-y.
Tomczak K, Czerwinska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015;19(1A):A68–77. https://doi.org/10.5114/wo.2014.47136.
Tarabichi M, Saiselet M, Tresallet C, et al. Revisiting the transcriptional analysis of primary tumours and associated nodal metastases with enhanced biological and statistical controls: application to thyroid cancer. Br J Cancer. 2015;112(10):1665–74. https://doi.org/10.1038/bjc.2014.665.
Zamanian-Azodi M, Rezaei-Tavirani M. Investigation of health benefits of cocoa in human colorectal cancer cell line, HT-29 through interactome analysis. Gastroenterol Hepatol Bed Bench. 2019;12(1):67–73.
Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–58. https://doi.org/10.1016/j.neo.2017.05.002.
Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 30: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9(2):166–80.
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013. https://doi.org/10.1126/scisignal.2004088.
Stelzer G, Rosen N, Plaschkes I, et al. The genecards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. 2016;54(1):1–30. https://doi.org/10.1002/cpbi.5.
Zhuo X, Chang A, Huang C, Yang L, Xiang Z, Zhou Y. Expression of TWIST, an inducer of epithelial-mesenchymal transition, in nasopharyngeal carcinoma and its clinical significance. Int J Clin Exp Pathol. 2014;7(12):8862–8.
Gheysarzadeh A, Bakhtiari H, Ansari A, Emami Razavi A, Emami MH, Mofid MR. The insulin-like growth factor binding protein-3 and its death receptor in pancreatic ductal adenocarcinoma poor prognosis. J Cell Physiol. 2019. https://doi.org/10.1002/jcp.28922.
Natsuizaka M, Kinugasa H, Kagawa S, et al. IGFBP3 promotes esophageal cancer growth by suppressing oxidative stress in hypoxic tumor microenvironment. Am J Cancer Res. 2014;4(1):29–41.
Chen CH, Chen PY, Lin YY et al. Suppression of tumor growth via IGFBP3 depletion as a potential treatment in glioma. J Neurosurg. 2019:1–12. https://doi.org/10.3171/2018.8.jns181217.
Wang YA, Sun Y, Palmer J, et al. IGFBP3 modulates lung tumorigenesis and cell growth through IGF1 signaling. Mol Cancer Res MCR. 2017;15(7):896–904. https://doi.org/10.1158/1541-7786.MCR-16-0390.
Han JJ, Xue DW, Han QR, et al. Induction of apoptosis by IGFBP3 overexpression in hepatocellular carcinoma cells. Asian Pac J Cancer Prevent APJCP. 2014;15(23):10085–9. https://doi.org/10.7314/apjcp.2014.15.23.10085.
Al-Daghri NM, Manousopoulou A, Alokail MS, et al. Sex-specific correlation of IGFBP-2 and IGFBP-3 with vitamin D status in adults with obesity: a cross-sectional serum proteomics study. Nutrit Diabet. 2018;8(1):54. https://doi.org/10.1038/s41387-018-0063-8.
Boker J, Volzke H, Nauck M, Hannemann A, Friedrich N. Associations of insulin-like growth factor-I and insulin-like growth factor binding protein-3 with bone quality in the general adult population. Clin Endocrinol. 2018;88(6):830–7. https://doi.org/10.1111/cen.13588.
Birzniece V, Magnusson NE, Ho KK, Frystyk J. Effects of raloxifene and estrogen on bioactive IGF1 in GH-deficient women. Eur J Endocrinol. 2014;170(3):375–83. https://doi.org/10.1530/EJE-13-0835.
Chen L, Gao H, Liang J, et al. miR-203a-3p promotes colorectal cancer proliferation and migration by targeting PDE4D. Am J Cancer Res. 2018;8(12):2387–401.
Yen YC, Hsiao JR, Jiang SS, et al. Insulin-like growth factor-independent insulin-like growth factor binding protein 3 promotes cell migration and lymph node metastasis of oral squamous cell carcinoma cells by requirement of integrin beta1. Oncotarget. 2015;6(39):41837–55. https://doi.org/10.18632/oncotarget.5995.
Wu JB, Tang YL, Liang XH. Targeting VEGF pathway to normalize the vasculature: an emerging insight in cancer therapy. OncoTargets Therapy. 2018;11:6901–9. https://doi.org/10.2147/OTT.S172042.
Fernandez-Aroca DM, Roche O, Sabater S, et al. P53 pathway is a major determinant in the radiosensitizing effect of Palbociclib: implication in cancer therapy. Cancer Lett. 2019;451:23–33. https://doi.org/10.1016/j.canlet.2019.02.049.
He XY, Pan XM, Jin MM, et al. Long non-coding RNA AK027294 promotes tumor growth by upregulating PCNA in gastric cancer. Eur Rev Med Pharmacol Sci. 2019;23(13):5762–9. https://doi.org/10.26355/eurrev_201907_18314.
Lu C, Huang T, Chen W, Lu H. GnRH participates in the self-renewal of A549-derived lung cancer stem-like cells through upregulation of the JNK signaling pathway. Oncol Rep. 2015;34(1):244–50. https://doi.org/10.3892/or.2015.3956.
Azrad M, Zhang K, Vollmer RT, et al. Prostatic alpha-linolenic acid (ALA) is positively associated with aggressive prostate cancer: a relationship which may depend on genetic variation in ALA metabolism. PLoS ONE. 2012;7(12):e53104. https://doi.org/10.1371/journal.pone.0053104.
Chiu AP, Tschida BR, Sham TT, et al. HBx-K130M/V131I promotes liver cancer in transgenic mice via AKT/FOXO1 signaling pathway and arachidonic acid metabolism. Mol Cancer Res MCR. 2019. https://doi.org/10.1158/1541-7786.MCR-18-1127.
Bouchal P, Dvorakova M, Roumeliotis T, et al. Combined proteomics and transcriptomics identifies carboxypeptidase B1 and nuclear factor kappaB (NF-kappaB) associated proteins as putative biomarkers of metastasis in low grade breast cancer. Mol Cell Proteomics. 2015;14(7):1814–30. https://doi.org/10.1074/mcp.M114.041335.
Acknowledgements
Not applicable.
Funding
The present study was partially supported by the National Natural Science Foundation of China (No. 81560298) and Special Foundation of China Postdoctoral Science (2015T80962).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of Guizhou Medical University and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individuals according to Shanghai Outdo Biotech Co., Ltd.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Huang, Y., Chang, A., Zhou, W. et al. IGFBP3 as an indicator of lymph node metastasis and unfavorable prognosis for papillary thyroid carcinoma. Clin Exp Med 20, 515–525 (2020). https://doi.org/10.1007/s10238-020-00642-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10238-020-00642-3