
Abstract
Advances in our understanding of genome structure provide consistent evidence for the existence of a core genome representing species classically defined by phenotype, as well as conditionally dispensable components of the genome that shows extensive variation between individuals of a given species. Generally, conservation of phenotypic features between species reflects conserved features of the genome; however, this is evidently not necessarily always the case as demonstrated by the analysis of the tunicate chordate Oikopleura dioica. In both plants and animals, the methylation activity of DNA and histones continues to present new variables for modifying (eventually) the phenotype of an organism and provides for structural variation that builds on the point mutations, rearrangements, indels, and amplification of retrotransposable elements traditionally considered. The translation of the advances in the structure/function analysis of the genome to industry is facilitated through the capture of research outputs in “toolboxes” that remain accessible in the public domain.
Similar content being viewed by others


Introduction
This mini-review is based on presentations and summaries presented at Plant and Animal Genome (PAG) XXI, in San Diego in January 2013. We note that where unpublished information is cited, permission to include the information in this manuscript was obtained from the presenters. The PAG meeting covers a very broad range of data exchange through many workshops and industry exhibitions, and the invited plenary lectures within a particular domain provide a particularly good snapshot of areas that are exciting and have relevance across the broader plant and animal disciplines. In the area of genome evolution and population level studies, plenary lectures were provided by Michele Morgante (University of Udine, Italy), Greger Larson (Durham University, UK) and Daniel Chourrout (Centre for Marine Biology, Norway); in the area of translating genomics to industry and modifying specific traits, Gary Muehlbauer (University of Minnesota, USA) provided insights into the cereals; in the area of analysing changes in gene expression of organs, Steven Jacobssen provided an extensive overview; in the area of informatics technology and communication, Eric Perakslis (US FDA, USA), Michael B Eisen (UC-Berkeley, USA) and Goncalo Abecasis (University of Michigan, USA) presented their views and achievements.
Analysing the basis for change in gene expression
The plenary lecture by Steven Jacobssen reviewed the status of epigenetic modification in chromatin and the general issue of gene silencing in plants. The methylation of cytosine (Cm, the “fifth base”) residues is now recognised as a major variable in the control of gene expression and assaying Cm can be carried out as a part of a genome sequencing project using bisulphite sequencing (Cokus et al. 2008). The Cm-containing genome sequences include CG, methylated by METHYLTRANSFERASE 1 (MET1); CHG, methylated by CHROMOMETHYLASE3 (CMT3); and CHH methylated by DOMAINS REARRANGED METHYLASE 2 (DRM2)Footnote 1. The DRM2 enzyme is required for the methylation of the cytosines in all the sequences. The methyltransferase, kryptonite (KYP/SUVH4), is responsible for the dimethylation of histone H3 and also binds to methylated DNA to provide an association between DNA and histone methylation that is similar to that observed in animals (Johnson et al. 2007). Specific regions of the genomic DNA such as retrotransposable elements can be methylated through an RNA directed pathway involving RNA polymerases IV and V, plus small interfering RNAs (Zhong et al. 2012). Demethylation of histones (Krichevsky et al. 2011) and specific loci in genomic DNA (Penterman et al. 2007) are additional variables in the control of gene expression. In a genome-wide analysis, Stroud et al. (2013) mapped the variation in methylation at the single nucleotide level within the Arabidopsis genome, in response to mutations in a set of 86 genes involved in the gene silencing pathway. As expected, mutation in MET1 eliminated CG methylation while mutations in the genes VARIANT IN METHYLATION (VIM1, VIM2 and VIM3) dramatically affected methylation but showed functional redundancy within the group. The gene DECREASE IN DNA METHYLATION 1 was associated with methylating mainly DNA in heterochromatin. The methylation of CHG, again as expected, was depleted by mutation in CMT3. Mutations in KYP, SUVH5 and SUVH6 showed similar effects when compared to each other. The mutations did not however show a uniform loss of CHG methylation and it was evident that the DRM1/2 genes also defined sites of methylation that did not overlap CMT3 targets. The methylation of the CHH motif was found to be closely linked to methylation of CHG but differentiation between the sites was found in that KYP SUVH5/6-regulated CHH methylation in a RNAi-dependent manner. The gene networks controlling DNA methylation included genes controlling the RNAi pathway as well as those modifying chromatin structure. It was evident that new factors and gene networks controlling DNA methylation remain to be discovered (Berdasco et al. 2008; Stroud et al. 2013).
Changes in DNA methylation in response to biotic stress (Dowen et al. 2012) and in the tissue culture of cells (Cheng et al. 2006) have been reported. In rice, the Tos17-LTR retrotransposable elements that are activated by the tissue culture process are undermethylated, in contrast to plants regenerated from tissue culture where the Tos17 elements are successively methylated again with each generation (Cheng et al. 2006). Loss of function of the SET DOMAIN GROUP PROTEIN (Baumbusch et al. 2001; Caro et al. 2012) in rice (SG714) decreases DNA methylation and increases the transposition of Tos17 elements (Ding et al. 2007). In the case of leaf cells of Arabidopsis reacting to the biotrophic pathogen Pseudomonas syringae, the fine mapping of changes in methylation of the genome (Dowen et al. 2012) has provided evidence for localised changes in methylation. A prominent phenotype is cell death in response to the pathogen. Although the overall distribution of Cm across the genome was similar before and after infection, a detailed analysis showed that changes in methylation occurred differentially in gene-rich regions compared to the rest of the genome with a peak in differentially methylated sites within 1 kb upstream from the start of transcription. A feature of the distribution was the differential methylation of transposable elements near protein coding genes (Dowen et al. 2012). The response of tissue to salicylic acid, rather than P. syringae, included more transposable elements and suggested that, on a broad scale, the programming of DNA methylation is integral to the control of gene expression.
Genome evolution and population level studies
Daniel Chourrout discussed the coastal marine planktonic chordate Oikopleura dioica in his plenary lecture. The genome and transcriptome resources for this organism are well established in the form of OikoBase (Danks et al. 2012) and early studies on ribosomal protein, EF-1a, Hox proteins and tubulin gene families (Edvardsen et al. 2004) indicated that these invertebrates have a very compact genome (18,020 predicted genes in 70 Mb, Denoeud et al. 2010) with short introns at variable positions within genes. The Hox genes, important in development, were not clustered as in most organisms studied to date (Seo et al. 2004). The genome is housed within three chromosomes plus an X and Y chromosome and has been found to be under-methylated compared to other species. The striking feature of O. dioica is that key elements of its genome, in an evolutionary context, are unique to the species even though its phenotype provides the basis for its unambiguous classification as a tunicate chordate (Stach et al. 2008). Most transposable superfamilies of retrotransposable elements are missing from the O. dioica genome and synteny in chromosomal gene order to organisms such as Amphioxus, Ciona, Caenorhabditis, and sea anemone that are related in phenotype has been lost (Denoeud et al. 2010). A striking contrast exists within the O. dioica genome between the basic compact structure of most of the genome and the structure of the Y chromosome which shows very large introns in the genes housed within this chromosome (Denoeud et al. 2010). The studies highlight the significant gaps in our understanding in relating genome structure to function/phenotype in an organism.
DNA repair genes are missing in the O. dioica genome which is consistent with the high mutation rates deduced from a comparison of the genome sequences from populations from the eastern Atlantic and eastern Pacific oceans (Denoeud et al. 2010). Mutations in non-silent sites of genes were low compared to changes in silent sites in genes, consistent with strong selection pressures (Denoeud et al. 2010). It appears that in the context of the core and conditionally dispensable regions of genome discussed below (following paragraph), the core elements of the O. dioica genome have changed extensively and that new DNA sequences have been recruited from the variable, conditionally dispensable, regions of the genome to provide the basis for retaining the evolutionary conserved phenotypes that characterise tunicate chordates.
Genome level of analysis of individuals within populations of higher eukaryotes has also modified the way genomes are perceived. In the human genome (PAG 2010 in Appels et al. 2010), the analyses to complete linear DNA sequences for each chromosome has demonstrated that a representative single reference genome sequence is not feasible and, instead, a more complex view is required to show INDEL polymorphisms, small inversions and duplications in order to provide a template for analysing SNPs. In his plenary lecture at the PAG, Michele Morgante discussed the pan-genome concept developed first in bacteria (Tettelin et al. 2005) as a basis for considering a genome as consisting of core and conditionally dispensable elements (Morgante et al. 2007). In bacteria, Tettelin et al. (2005) compared the genome sequences of six strains of Streptococcus agalactiae to those available in databases and found that 80 % of the genome sequences could be assigned to a core genome. The remaining 20 % (referred to as dispensable) was found to be highly variable and included strain-specific genes, and was suggested to provide a reservoir of genes for modifying the pan-genome. Mobile and extrachromosomal elements were found to be prominent in the dispensable part of the genome. Based on the frequency of discovery of unique genes in this dispensable part of the genome, Tettelin et al. (2005) suggested that the total gene complement of a species may in fact be difficult to determine because new genes can be continually acquired into this part of the genome through lateral gene transfer.
In eukaryotes, the properties of the conditionally dispensable elements of the genome were classically illustrated in the control of gene expression by heterochromatin in Drosophila (Hilliker and Appels 1982). Regions of chromosomes defined, by cytology, as heterochromatin contain long intergenic tracts of simple sequence repeats and can moderate gene expression depending on their position within the genome (Hilliker and Appels 1982). The long tracts of gene-free genome sequences are not actually essential for life as demonstrated experimentally by generating large deletions in mice (Nobrega et al. 2004) and can therefore be defined as conditionally dispensable. A property of conditionally dispensable parts of the genome noted by Morgante would include the co-option of DNA sequences which contribute to differentiating individuals within a species. These regions of the genome that differentiate individuals could also contribute to heterosis/hybrid vigour (reviewed in Springer and Stupar 2007a) since hybrids show non-additive gene expression, outside the range of the parents. If the elements and strain-specific genes within conditionally dispensable regions of the genome contribute to heterosis through processes of complementation, it would be as part of a suite of molecular mechanisms ranging from new protein–protein interactions to new epigenetic states within the hybrids (Springer and Stupar 2007a, b).
The database of DNA sequence information from the genomes of plants and animals is rapidly expanding and in his plenary lecture Morgante showed how this provides a detailed view of the conditionally dispensable parts of the genome focused on the analysis of Zea mays (maize) and Vitis vinifera (grape) genomes. The whole genome comparison between the maize lines Mo17 and B73 (Brunner et al. 2005; Morgante et al. 2005) provided evidence for extensive differences between the two genomes driven by changes in the retrotransposable element component of the genome, even though crosses between lines are fertile. The regions of the genome not shared between the lines amounted to 50 % of the total sequence space. The fragments of genes in these highly polymorphic regions included sequences located in helitron transposable elements that were distributed among the different maize chromosomes, as determined from analysing oat–maize chromosome addition lines (Morgante et al. 2005). Regions of gene clusters tended to be restricted in their distribution within the genome compared to single exons. The more detailed comparison of specific loci on chromosomes 1S, 1L, 2S, 7S and 9S by Brunner et al. (2005) between Mo17 and B73 showed that colinearity between the genome regions was fragmented due to the insertion of long terminal repeat (LTR) retrotransposons and gene fragments. The age of these inserted LTRs was more recent than that of the colinear parts of the genome. The effective population size of the non-colinear or dispensable, sequence segments have been noted to differ from that of the core genome regions (Brunner et al. 2005) and where they are linked closely to genes affecting agronomically important traits would be expected to have significant effects on breeding. In addition to qualitative differences between related chromosome regions, quantitative differences in sequences >1 kb in size (CNVs) relative to B73, for 13 maize lines (including Mo17), have been determined using an array-based comparative genome hybridisation approach (Belo et al. 2010a). Approximately half of the 2,109 dispersed and clustered CNVs occurred in only one of the maize lines and have been considered to be potential contributors to heterosis (Belo et al. 2010b).
The advances in the DNA sequence-based characterization of the 14 varieties of grapes discussed by Morgante was aided by the availability of an extensive description of the grape genome (Jaillon et al. 2007; http://www.genoscope.cns.fr/externe/GenomeBrowser/Vitis/). The 19 Vitis chromosomes are represented today by a genome assembly of 33 mapped ultracontigs (N50 = 23 Mb) that provide a 91.2 % coverage of the genome. Historically, the domestication and development of grape varieties is closely linked to human settlement and agricultural practises, from the southern borders of the Black and Caspian seas to Afghanistan, ca 4000 BC (Olmo 1976). The transfer of grape plants to environments beyond this natural range led to hybridisation to wild Vitis species and produced new lines better adapted to the local environment (Cipriani et al. 2010). It is estimated that 10,000 varieties derive from the V. vinifera species originally domesticated in the Middle East (Olmo 1976). Genetic heterozygosity is a feature of this crop (Scalabrin et al. 2011). The resequencing analysis of the 14 grape varieties reported by Morgante used paired end sequencing and software such as BreakDancer (Chen et al. 2009) and DNACopy (Venkatraman and Olshen 2007), for analysing structural variation between the genome sequences. A total of 36,000 INDELS, 1–25 kb in length, were identified and it was deduced from the analyses that these were mainly due to transposition events resulting from the movement of Copia and Gypsy elements. The LINE elements were particularly polymorphic and it was found that these polymorphic elements were prominent in introns. An additional 147 Mbp of large deletions were identified with a depth of coverage approach. The genes in the stilbene (Vannozzi et al. 2012), terpene (Martin et al. 2010) and flavonoid pathways (Falginella et al. 2010) were used to illustrate the variation found. In the anthocyanin pathway, Kobayashi et al. (2004) for example demonstrated that a retrotransposon (Gret1, 10,422 bp) insertion into the promoter region of the transcription factor VvmybA1, was associated with the loss of pigmentation in white cultivars of V. vinifera. This characterization of the red vs white skin colour of grapes was consistent with the importance of transposition events in the domestication of grapes. A recent transcriptome analysis of V. vinifera cv Corvina (Venturini et al. 2013) identified 180 genes in the conditionally dispensable space of the genome, with 50 being differentially expressed.
The role of introgression to build up the conditionally dispensable regions of genomes, in the way indicated above for maize and grapes, throughout the history of domestication was discussed in detail by Greger Larson. The availability of a large, and rapidly expanding, databases of plant and animal species used as a food source provide a basis for defining the role of introgression in the history of domestication. In particular, Larson discussed the use of the DNA sequence databases to quantify variation and estimate the start of the domestication process while accepting that the process does not have an end and continues to this day (Larson and Burger 2013). Three separate domestication pathways have been defined by Zeder (2012) and have facilitated the development of population models that define the duration of the early capture period, the presence and size of bottlenecks, and the number and geographic distribution of potential ancestral populations. In the “commensal pathway”, a close link to the advent of agriculture is usually evident followed by increasing degrees of deliberate human action as the relationship between humans and the respective plants and animals increased (Larson and Burger 2013). Animals that were initially predated upon by people are included in the “prey pathway” where animals that were first hunted away from human settlements were subsequently more directly managed as they were brought into closer proximity with people. The population structure most often indicates a major bottleneck and short time frame for change. Plants and animals that followed the “directed pathway” tended to do so after a long time within the agriculture system and the process bypasses the early phases of habituation and management and begins with the collection of plants and the capture of wild animals with the deliberate intention of controlling their breeding. This directed pathway is accompanied by a dramatic bottleneck.
In the commensal and prey pathways, admixtures of populations are an important feature of the domestication process. The establishment of a high quality (BAC-based) genome reference sequence for pigs (Groenen et al. 2012) has allowed the analysis of the features of their domestication. Studies using nuclear DNA sequences (Groenen et al. 2012) and mitochondrial DNA sequences from both ancient DNA and present-day samples by Larson et al. (2010) have provided evidence consistent for the early evolution of Sus scrofa in the Island South East Asia (ISEA) region followed by migration into the rest of the Asia region and Eurasia. The Chinese domestic pig was argued to be a direct descendant from this ISEA centre of diversity (Larson et al. 2010). Evidence for other centres of domestication in the Indo-Burma and mountainous South East Asia regions (Larson et al. 2010; Charoensook et al. 2011) and a number of centres in Eurasia (Groenen et al. 2012) form the basis for the network of domestication. Bottlenecks in the pig lineages due to climatic change also need to be considered together with extinction events such as that of a Pacific clade haplotype in South East Asia (Larson et al. 2010). Admixture analyses carried out by Groenen et al. (2012) on nuclear DNA sequences within S. scrofa lineages demonstrated gene flow between the northern Chinese and European populations and varying degrees of exchange between domesticated pig lineages and their wild relatives. The latter was consistent with the semi-managed state in which pigs were kept in the early agricultural communities (Groenen et al. 2012).
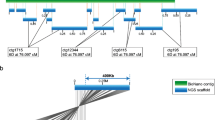
The analyses of other species closely associated with human societies (Larson et al. 2012; Larson and Burger 2013) consistently indicate that initial domestication processes are clearly distinguishable from subsequent movement and admixture with local wild populations. It was also evident that large-scale genome sequence analyses will continue to impact on defining the details of the co-evolution of human societies and groups of plants and animals. In rice and other cereals, the extensive genome sequencing is leading to better utilisation of wild relatives for crop improvement even though comparisons between the different Oryza genomes (for example) have demonstrated extensive DNA rearrangement in the colinearity of genes (Tian et al. 2011). Comparisons between the D genome donor to bread wheat and barley show conservation of gene order (Fig. 1) against a background of structural variation that is known to exist. For both rice and bread wheat, the wild relatives provide an extensive array of new gene alleles, and genes per se, for cultivar improvement (Kovach and McCouch 2008; Trethowan and Mujeeb-Kazi 2008; Appels et al. 2012) in the directed pathway category of the domestication process discussed by Larson.

The advances in cereal genome sequencing provides the basis for the alignment of genomes and the identification of candidate genes underpinning significant agronomic traits. The alignment of the barley and D genome donor of wheat (Aegilops tauschii) genome sequences is based on the genes that are conserved between the species and illustrates the high degree of synteny between the barley and wheat genomes that can now be exploited for studying specific agronomic traits in detail. The image was kindly provided by M Pfeifer and K Mayer (MIPS, Helmholtz-Zentrum Munich, Germany), and for a more extensive comparison between the D genome and barley, see Brenchley et al. (2012) and Jia et al. (2013)
Translation of genomics to industry and modifying specific traits
Genome sequencing of cereals is now maturing as the BAC-based sequence assemblies for rice (rgp.dna.affrc.go.jp/IRGSP), maize (www.maizegdb.org) and barley (International Barley Genome Sequencing Consortium, IBGSC 2012) are being utilised across a broad range of applications, with key outputs being the projection of phenotypic traits important to the industry onto the genome DNA sequence. For the hexaploid wheat (IWGSC—www.wheatgenome.org; Brenchley et al. 2012) and the A genome (Ling et al. 2013) and D genome (Jia et al. 2013; see also Fig. 1) diploid genome donors, the whole genome sequencing technology has provided an important step in assigning genome sequences to molecular genetic maps and traits of agronomic significance (Feuillet et al. 2012). In his plenary presentation, Gary Muehlbauer provided a focus on barley molecular genetics with particular reference to Fusarium head blight (FHB) resistance and integrating genomics resources and genetic variation in wild barleys into barley breeding programmes. The current physical/sequence map for barley comprises 4.98 Gb with 79,379 transcript clusters identified through alignments with cDNA and RNA-seq data. The annotated genes include 26,159 genes that were supported by homology to genes in other plant genomes (for example see Fig. 1; Middleton et al. 2012). Based on RNA-seq data, 55 % of the genes classified as high confidence showed evidence for alternative splicing (IBGSC 2012) and this was argued to represent a significant variable in linking gene expression to the final phenotype. The retrotransposable element/repetitive sequence content of the genome was estimated to represent 84 % of the total DNA sequence. In addition to the mapping of single nucleotide polymorphisms by sequencing (IBGSC 2012), surveys of wild and cultivated barleys by comparative genome hybridisation arrays (Muñoz-Amatriaín et al. 2010) have also demonstrated that a significant proportion (15 %) of the barley genome is affected by copy number variation (CNV) in DNA sequences. The telomeric regions of chromosomes were enriched for CNVs and correlated with an enrichment of genes in these regions. It is possible that the CNVs contribute to the phenotypic diversity of barleys (Muñoz-Amatriaín et al. 2010) and may relate to the conditionally dispensable regions of the genome discussed earlier.
In order to translate the basic findings of the barley genome into industry, Muehlbauer argued that contemporary breeders utilised molecular data to predict phenotypes that are of value in novel germplasm for increasing the speed and efficiency of their programmes as well as reducing costs (Waugh et al. 2010; Comadran et al. 2011; Okagaki et al. 2012; Blake et al. 2012; Berger et al. 2012). The outputs from research and germplasm analysis projects in barley are made available to the industry through The Hordeum Toolbox (THT) which integrates extensive phenotypic and genotypic data sets for further downstream analyses (Szűcs et al. 2009; Blake et al. 2012). THT was argued to facilitate the sharing of data between breeding programmes. The analysis of a complex trait such FHB has necessitated the analysis of a broad range of variables ranging from defining resistance haplotypes in wild germplasms through to defining QTL for FHB resistance on chromosome 2H and 6H in new molecular marker genetic maps (Huang et al. 2013). The analysis uncoupled FHB resistance from head phenotype (two rows vs six rows, Cuesta-Marcos et al. 2010) and identified novel alleles for the known and robust FHB QTL and provided a good example for the broad integration of technologies. Progressive use of Genome Wide Association Studies (GWAS, Berger et al. 2013; Cuesta-Marco et al. 2010) expands the database of molecular information linked to phenotype and THT provides a valuable model for translating these data to industry.
Informatics technologies and communication
The plenary lecture by Goncalo Abecasis provided insights into the computational analysis of complex disease loci and finding rare variants associated with disease in human populations. The ability to attribute or impute genome variation to a particular disease has improved as the data set of genome sequences has increased (The 1000 Genomes Project Consortium 2012). A specific example was discussed related to macular degeneration (Li et al. 2006b; Rachauduri et al. 2011) and its association with a ca 70 kb deletion on the long arm of chromosome 1 (CFHR1-CFHR3) and SNPs in a closely linked complement factor H locus (CFH). Overall, the analysis indicated that the CFHR1-CFHR3 deletion and CFH locus accounted for “modest ” levels of variation in the phenotype, consistent with the possibility of multiple susceptibility alleles in the region of chromosome 1 that was analysed (Li et al. 2006a, b). In general, it is apparent that integrating additional knowledge related to the ancestry of individuals analysed and biological attributes of the trait studied (Li et al. 2006a; Chasman et al. 2012) is important in utilising GWAS for defining loci contributing to complex traits. In addition, DNA sequence information from more individuals would increase the power of detecting rare alleles. The challenge of attaining a balance between cost of sequencing with respect to genome coverage, genotype calling and statistical power of the output data for GWAS was discussed by Abecasis from the computation point-of-view through the use of the AbCD (Kang et al. 2013) and Triocaller (Chen et al. 2013) software.
The plenary lectures by Eric Perakslis and Michael Eisen debated different aspects of freedom to access information for scientific research. In the Food and Drug Administration of the USA, Perakslis described the large scale of the records that are kept in relation to food and drug imports and the use of PREDICT software (www.fda.gov/Forindustry/Importprogram/ucm172743) to handle the risk-based screening procedures and anticipating innovation. The database underpinning PREDICT required the details of contents including points of origin of the different components within food and drug products and was a potentially valuable resource for pre-competitive research related to food security. It was noted by Perakslis that this level of data sharing required an “honest broker” within the frame work of the Federal Information Security Management Act. The issue of data sharing and data access was also addressed by Michael Eisen. The focus for Eisen was the contrasting situations that related to DNA, RNA and protein sequence databases which were free to access and the access to scientific text in the form of publications that was generally only accessible after payment of a fee. The argument was made that in an area of science such as represented by the PAG, sophisticated search engines equivalent to BLAST for DNA, RNA and protein sequences should be available for words/concepts in order to enrich the interpretation of complex data sets and draw on observations and conclusions from the analysis of organisms outside a particular focused area of interest.
Notes
The H designation represents the bases A, C or T
References
Appels R, Barrero R, Bellgard M (2012) Advances in biotechnology and linking outputs to variation in complex traits: plant and Animal Genome meeting January 2012. Funct Integr Genomics 12:1–9
Appels R, Barrerro R, Keeble G, Bellgard M (2010) Advances in genome studies: the PAG 2010 conference. Funct Integr Genomics 10:1–9
Baumbusch LO, Thorstensen T, Krauss V, Fischer A, Naumann K, Assalkhou R, Schulz I, Reuter G, Aalen RB (2001) The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res 29:21
Belo A, Beatty MK, Hondred D, Fengler KA, Bailin Li B, Rafalski A (2010a) Allelic genome structural variations in maize detected by array comparative genome hybridization. Theor Appl Genet 120:355–367
Belo et al (2010b) TAG. High resolution mapping of genomic structural variation. Nat Methods 6:677–681
Berdasco M, Alcazar R, Garcia-Ortiz MV, Ballestar E, Fernandez AF, Roldan-Arjona T, Tiburcio AF, Altabella T, Buisine N, Quesneville H, Baudry A, Lepiniec L, Alaminos M, Rodriguez R, Lloyd A, Colot V, Bender J, Canal MJ, Esteller M, Fraga MF (2008) Promoter DNA hypermethylation and gene repression in undifferentiated Arabidopsis cells. PLoS One 3:e3306
Berger GL, Liu S, Hall MD, Brooks WS, Chao S, Muehlbauer GJ, Baik BK, Brian Steffenson B, Griffey CA (2013) Marker-trait associations in Virginia Tech winter barley identified using genome-wide mapping. Theor Appl Genet 126:693–710
Berger GL, Liu S, Hall MD, Brooks WS, Chao S, Muehlbauer GJ, Baik BK, Steffenson B, Griffey CA (2012) Marker-trait associations in Virginia Tech winter barley identified using genome-wide mapping. Theor Appl Genet 126(3):693–710
Blake VC, Kling JG, Hayes PM, Jannink J-L, Jillella SR, Lee J, Matthews DE, Chao S, Close TJ, Muehlbauer GJ, Smith KP, Wise RP, Dickerson JA (2012) The Hordeum Toolbox: the barley coordinated agricultural project genotype and phenotype resource. Plant Genome 5:81–91
Brenchley R, Spannagl M, Pfeifer M, Barker GLA, D’Amore R, Allen AM, McKenzie N, Kramer M, Kerhornou A, Bolser D, Kay S, Waite D, Trick M, Bancroft I, Gu Y, Huo N, Luo M-C, Sehgal S, Gill BS, Kianian S, Anderson O, Kersey P, Dvorak J, McCombie WR, Hall A, Mayer KFX, Edwards KJ, Bevan MW, Hall N (2012) Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491:705–710
Brunner S, Fengler K, Morgante M, Tingey S, Rafalski A (2005) Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 17:343–360
Caro E, Stroud H, Greenberg MVC, Bernatavichute YV, Suhua Feng S, Groth M, Vashisht AA, Wohlschlegel J, Jacobsen SE (2012) The SET-domain protein suvr5 mediates h3k9me2 deposition and silencing at stimulus response genes in a DNA methylation-independent manner. PLoS Genet 8(10):e1002995
Charoensook R, Brenig B, Gatphayak K, Knorr C (2011) Further resolution of porcine phylogeny in Southeast Asia by Thai mtDNA haplotypes. Anim Genet 42:445–450
Chasman DI et al (2012) Integration of genome-wide association studies with biological knowledge identifies six novel genes related to kidney function. Hum Mol Genet 21:5329–5343
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, Shi X, Fulton RS, Ley TJ, Wilson RK, Ding L, Mardis ER (2009) BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6:677–681
Chen W, Li B, Zeng Z, Sanna S, Sidore C, Busonero F, Kang HM, Li Y, Abecasis GR (2013) Genotype calling and haplotyping in parent–offspring trios. Genome Res 23:142–151
Cheng C, Daigen M, Hirochika H (2006) Epigenetic regulation of the rice retrotransposon Tos17. Mol Gen Genomics 276:378–390
Cipriani G, Spadotto A, Jurman I, Di Gaspero G, Crespan M, Meneghetti S, Frare E, Vignani R, Cresti M, Morgante M, Pezzotti M, Pe E, Policriti A, Testolin R (2010) The SSR-based molecular profile of 1005 grapevine (Vitis vinifera L.) accessions uncovers new synonymy and parentages, and reveals a large admixture amongst varieties of different geographic origin. Theor Appl Genet 121:1569–1585
Cokus SJ, Feng S, Zhang X, Chen Z, Barry Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Vol 452
Comadran J, Ramsay L, MacKenzie K, Hayes P, Close TJ, Muehlbauer G, Stein N, Waugh R (2011) Patterns of polymorphism and linkage disequilibrium in cultivated barley. Theor Appl Genet 122:523–531
Cuesta-Marco A, Szűcs P, Close TJ, Filichkin T, Muehlbauer GJ, Smith KP, Hayes PM (2010) Genome-wide SNPs and re-sequencing of growth habit and inflorescence genes in barley: implications for association mapping in germplasm arrays varying in size and structure. BMC Genomics 11:707
Danks G, Campsteijn CC, Parida M, Butcher S, Doddapaneni H, Fu B, Petrin R, Metpally R, Lenhard B, Wincker P, Chourrout D, Thompson EM, Manak JR (2012) OikoBase: a genomics and developmental transcriptomics resource for the urochordate Oikopleura dioica. Nucl Acids Res 41:D845–D853
Denoeud F et al (2010) Plasticity of animal genome architecture unmasked by rapid evolution of a pelagic tunicate. Science 330:1381–1385
Ding Y, Wang X, Su LS, Zhai JX, Cao SY, Zhang DF, Liu CY, Bi YP, Qian Q, Cheng ZK, Chu CC, Cao XF (2007) SDG714, a histone H3K9 methyltransferase, is involved in Tos17 DNA methylation and transposition in rice. Plant Cell 19:9–22
Dowen RH, Pelizzola M, Schmitz RJ, Ryan Lister R, Dowen JM, Nery JR, Dixon JE, Ecker JR (2012) Widespread dynamic DNA methylation in response to biotic stress. Proc Natl Acad Sci U S A 109(32):E2183–E2191
Edvardsen RB, Lerat E, Dorthea A, Maeland M, Flat M, Tewari R, Jensen MF, Lehrach H, Reinhardt R, Seo HC, Chourrout D (2004) Hypervariable and highly divergent intron–exon organizations in the chordate Oikopleura dioica. J Mol Evol 59:448–457
Falginella L, Castellarin SD, Testolin R, Gambetta GA, Morgante M, Di Gaspero G (2010) Expansion and subfunctionalisation of flavonoid 3′,5′-hydroxylases in the grapevine lineage. BMC Genomics 11:562
Feuillet C et al (2012) Integrating cereal genomics to support innovation in the Triticeae. Funct Integr Genomics 12(4):573–583
Groenen MA, Swine Genome Sequencing Consortium (2012) Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491:393–398
Hilliker AJ, Appels R (1982) Pleiotropic effects associated with the deletion of heterochromatin surrounding rDNA on the X chromosome of Drosophila. Chromosoma (Berl) 86:469–490
Huang Y, Millett BP, Beaubien KA, Dahl SK, Steffenson BJ, Smith KP, Muehlbauer GJ (2013) Haplotype diversity and population structure in cultivated and wild barley evaluated for Fusarium head blight responses. Theor Appl Genet 126:619–636
International Barley Genome Sequencing Consortium et al (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491:711–716
Jaillon O, French-Italian Public Consortium for Grapevine Genome Characterization et al (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449(7161):463–467
Jia J, Shancen Zhao S, Kong X, Li Y, Zhao G, He W, Appels R, Pfeifer M, Tao Y, Zhang X, Jing R, Zhang C, Ma Y, Gao L, Gao C, Spannagl M, Mayer KFX, Dong Li D, Pan S, Fengya Zheng F, Hu Q, Xia X, Li J, Liang Q, Chen J, Wicker T, Gou C, Kuang H, He G, Luo Y, Keller B, Xia Q, Lu P, Wang J, Zou H, Zhang R, Gao J, Middleton C, Quan Z, Liu G, Wang J, IWGSC, Yang H, Xu Liu X, He Z, Mao L Wang J. (2013) The Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature. doi:1038/nature12028
Johnson LM, Bostick M, Zhang X, Kraft E, Henderson I, Judy Callis J, Jacobsen SE (2007) The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr Biol 17:379–384
Kang J, Huang KC, Xu Z, Wang Y, Abecasis GR, Li Y. (2013) AbCD: arbitrary coverage design for sequencing-based genetic studies. Bioinformatics. doi:10.1093/bioinformatics/btt041
Kobayashi S, Goto-Yamamoto N, Hirochika H (2004) Retrotransposon-induced mutations in grape skin color. Science 304:982
Kovach MJ, McCouch SR (2008) Leveraging natural diversity: back through the bottleneck. Curr Opin Plant Biol 11:193–200
Krichevsky A, Adi Zaltsman A, Lacroix B, Citovsky V (2011) Involvement of KDM1C histone demethylase-OTLD1 otubain-like histone deubiquitinase complexes in plant gene repression. Proc Natl Acad Sci U S A 108:11157–11162
Larson G, Burger J (2013). A population genetic view of animal domestication. Trends in Genetics. doi:10.1016/j.tig.2013.01.003
Larson G, Karlsson EK, Perri A, Webster MT, Ho SYW, Peters J, Stahl PW, Piper PJ, Lingaas F, Fredholm M, Comstock KE, Modiano JF, Schelling C, Agoulnik AI, Leegwater PA, Dobney K, Vigne J-D, Vila C, Andersson L, Lindblad-Toh K (2012) Rethinking dog domestication by integrating genetics, archeology, and biogeography. Proc Natl Acad Sci U S A 109:8878–8883
Larson G et al (2010) Patterns of East Asian pig domestication, migration, and turnover revealed by modern and ancient DNA. Proc Natl Acad Sci USA 107:7686–7691
Li B, Chen W, Zhan X, Busonero F, Sanna S, Carlo Sidore C, Cucca F, Kang HM, Abecasis GR (2006a) A likelihood based framework for variant calling and de novo mutation detection in families. PLoS Genet 8:e1002944
Li M, Atmaca-Sonmez P, Othman M, Branham KE, Khanna R, Wade MS, Li Y, Liang L, Zareparsi S, Swaroop A, Abecasis GR (2006b) CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nat Genet 38:1049–1054
Ling H-Q, Zhao S, Liu D, Wang J, Sun H, Zhang C, Fan H, Li D, Dong L, Tao Y, Gao C, Wu H, Li Y, Cui Y, Xiaosen Guo X, Zheng S, Wang B, Yu K, Liang Q, Wenlong Yang W, Lou X, Chen J, Feng M, Jian J, Zhang X, Luo G, Jiang Y, Junjie Liu J, Wang Z, Sha Y, Zhang B, Huajun Wu H, Dingzhong Tang D, Shen Q, Xue P, Zou S, Wang X, Liu X, Wang F, Yang Y, Xueli An X, Dong Z, Zhang K, Xiangqi Zhang X, Luo M-C, Dvorak J, Tong Y, Wang J, Yang H, Li Z, Wang D, Zhang A, Wang J. (2013) The Draft Genome of Triticum urartu 1—the progenitor of the wheat A genome. Nature. doi:105524/100050
Martin DM et al (2010) Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol 10:226
Middleton CP, Stein N, Keller B, Kilian B, Wicker T (2012) Comparative analysis of genome composition in Triticeae reveals strong variation in transposable element dynamics and nucleotide diversity. Plant J. doi:10.1111/tpj.12048
Morgante M, Brunner S, Pea G, Fengler K, Zuccolo A, Rafalski A (2005) Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat Genet 37:997–1002
Morgante M, De Paoli E, Radovic S (2007) Transposable elements and the plant pan-genomes. Curr Opin Plant Biol 10:149–155
Muñoz-Amatriaín M, Xiong Y, Schmitt MR, Bilgic H, Budde AD, Chao S, Smith KP, Muehlbauer GJ (2010) Transcriptome analysis of a barley breeding program examines gene expression diversity and reveals target genes for malting quality improvement. BMC Genomics 11:653
Nobrega MA, Zhu Y, Plajzer-Frick I, Afzal V, Rubin EM (2004) Megabase deletions of gene deserts result in viable mice. Nature 431:988–993
Okagaki RJ, Cho S, Kruger WM, Xu WW, Heinen S, Muehlbauer GJ. (2012) The barley UNICULM2 gene resides in a centromeric region and may be associated with signaling and stress responses. Funct Integr Genomics. doi:10.1007/s10142-012-0299-7
Olmo HP (1976) Grapes: Vitis and Muscadinia. In: Simmonds NW (ed) Evolution of crop plants. Longman, London, pp 294–298
Penterman J, Uzawa R, Fischer RL (2007) Genetic interactions between DNA demethylation and methylation in Arabidopsis. Plant Physiol 145:1549–1557
Rachauduri S, Iartchouk O, Chin K, Tan P, Tai A, Ripke S, Gowrisankar S, Vemuri S, Montgomery K, Yu Y, Reynolds R, Zack DJ, Campochiaro P, Katsanis N, Daly M, Seddon JM (2011) A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet 43:1232–1238
Scalabrin S, Troggio M, Moroldo M, Pindo M, Felice N, Coppola G, Prete G, Malacarne G, Marconi R, Faes G, Jurman I, Grando S, Jesse T, Segala C, Valle G, Policriti A, Fontana P, Morgante M, Velasco R (2011) Physical mapping in highly heterozygous genomes: a physical contig map of the Pinot Noir grapevine cultivar. BMC Genomics 11:204
Seo HC, Edvardsen RB, Maeland AD, Bjordal M, Jensen MF, Hanssen A, Flaat M, Weissenbach J, Lehrach H, Wincker P, Reinhardt R, Chourroutt D (2004) Hox cluster disintegration with persistent anteroposterior order of expression in Oikopleura dioica. Nature 413:67–71
Springer NM, Stupar RM (2007a) Allelic variation and heterosis in maize: how do two halves make more than a whole? Genome Res 17:264–275
Springer NM, Stupar RM (2007b) Allele-specific expression patterns reveal biases and embryo-specific parent-of-origin effects in hybrid maize. Plant Cell 19:2391–2402
Stach T, Jonas Winter J, Bouquet J-M, Chourrout D, Schnabel R (2008) Embryology of a planktonic tunicate reveals traces of sessility. Proc Natl Acad Sci U S A 105:7229–7234
Stroud H, Greenberg MVC, Feng S, Bernatavichute YV, Jacobsen SE (2013) Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152:352–364
Szűcs P, Blake VC, Bhat PR, Chao S, Close TJ, Cuesta-Marcos A, Muehlbauer GJ, Ramsay L, Waugh R, Hayes PM (2009) An integrated resource for barley linkage map and malting quality QTL alignment. Plant Genome 2:134–140
Tettelin H et al (2005) Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci 102:13950–13956
The 1000 Genomes Project Consortium (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65
Tian Z, Yu Y, Lin F, Yu Y, Sanmiguel PJ, Wing RA, McCouch SR, Ma J, Jackson SA (2011) Exceptional lability of a genomic complex in rice and its close relatives revealed by interspecific and intraspecific comparison and population analysis. BMC Genomics 12:142–154
Trethowan RM, Mujeeb-Kazi A (2008) A novel germplasm resources for improving environmental stress tolerance of hexaploid wheat. Crop Sci 48:1255–1265
Vannozzi A, Dry IB, Fasoli M, Zenoni S, Lucchin M (2012) Genome-wide analysis of the grapevine stilbene synthase multigenic family: genomic organization and expression profiles upon biotic and abiotic stresses. BMC Plant Biol 12:130
Venkatraman E, Olshen A (2007) A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23:657–663
Venturini L, Ferrarini A, Sara Zenoni S, Tornielli GB, Fasoli M, Dal Santo S, Minio A, Buson G, Tononi P, Zago ED, Zamperin G, Bellin D, Pezzotti M, Delledonne M (2013) De novo transcriptome characterization of Vitis vinifera cv. Corvina unveils varietal diversity. BMC Genomics 14:41
Waugh R et al (2010) Whole-genome association mapping in elite inbred crop varieties. Genome 53:967–972
Zeder MA (2012) The domestication of animals. J Anthropol Res 68:161–190
Zhong X, Hale CJ, Law JA, Johnson LM, Feng S, Tu A, Jacobsen SE (2012) DDR complex facilitates global association of RNA polymerase V to promoters and evolutionarily young transposons. Nat Struct Mol Biol 19:870–875
Acknowledgments
The authors acknowledge the organisers of the Plant and Animal Genome for bringing together a stimulating conference in January each year and that the content of this meeting serves to provide a snapshot of developments in the genomics area as a basis for this mini-review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Appels, R., Barrero, R. & Bellgard, M. Advances in biotechnology and informatics to link variation in the genome to phenotypes in plants and animals. Funct Integr Genomics 13, 1–9 (2013). https://doi.org/10.1007/s10142-013-0319-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-013-0319-2