
Abstract
Introduction
Because spermatocytic tumors of the testis are rare, only limited evidence exists regarding the malignant potential and the optimal management of localized and metastatic disease.
Materials and methods
We performed a systematic review through MEDLINE, EMBASE, Scopus, Cochrane Database of Systematic Reviews and Web of Science to identify reports including patients with testicular spermatocytic tumors.
Results
From originally 7863 studies, we extracted data of 146 patients of which 99% were treated with radical orchiectomy. Metastases in patients with initially localised disease were diagnosed in 7% of patients and detected after a median follow-up of 5.5 months (range 2–21 months). Patients with aggressive histology (sarcoma or anaplastic subtype) were more likely to have metastatic disease (6/124 (5%) vs 9/22 (41%), p < 0.001). Patients with metastatic disease had larger primary tumors (92.5 vs 67.5 mm, p = 0.05). Life expectancy in patients with metastatic disease ranged from 1 to 25 months.
Conclusion
The published literature does neither support the use of testis sparing surgery nor adjuvant therapy. Patients with aggressive variants or larger tumors were more likely to have metastases and develop recurrences within the first few years. Patients with metastatic disease have a limited life expectancy and metastatic spermatocytic tumors are not as responsive to chemotherapy as germ cell cancers.
Similar content being viewed by others


Introduction
Spermatocytic tumors are extremely rare and occur exclusively in the testis. Due to a morphological overlap to classic seminoma, they were named as “spermatocytic seminomas” in the past. However, the most recent World Health Organization (WHO) Classification of Tumors of the Urinary System and Male Genital Organs distinguished between spermatocytic tumors and seminomas because of several important differences (Moch et al. 2016). First, spermatocytic tumors are not derived from germ cell neoplasia in situ and lack 12p alterations (Rosenberg et al. 1998). Second, spermatocytic tumors show a unique amplification of chromosome 9 corresponding to the DMRT1 gene and are never associated with other forms of germ cell tumors (Looijenga et al. 2006). Third, seminomas/dysgerminomas originate from primordial germ cells or gonocytes expressing stem cell-specific genes (e.g., POU5F1, PROM1/CD133, and ZFP42), whereas spermatocytic tumors originate from primary spermatocytes and that have at least initiated prophase meiosis (expressing TCFL5, CLGN, and LDHc) (Looijenga et al. 2006).
Most spermatocytic tumors of the testis are considered benign tumors. As spermatocytic tumors are extremely rare, there is paucity of data on the optimal management of patients with localized or metastatic disease. It is unclear if testis sparing surgery (TSS) is sufficient, or if radical orchiectomy should be performed, if adjuvant treatment options may decrease relapse rates, which therapy should be recommended in the metastatic setting and how patients should be followed-up. We conducted a systematic literature review to identify and summarize all information on the clinical presentation, clinicopathological risk factors, available treatments, and survival information of patients with spermatocytic tumors.
Materials and methods
Data acquisition and search strategy
This review was performed according to Preferred Reporting Items for Systematic Reviews (PRISMA) statement (Moher et al. 2009, 2015). The review protocol was published in PROSPERO database (http://www.crd.york.ac.uk/PROSPERO; Registration number CRD42018108563).
A literature search was conducted using the electronic databases MEDLINE, EMBASE, Scopus, Cochrane Database of Systematic Reviews and Web of Science up to May 5th, 2018. After discussion, a clinical medical librarian applied a broad approach using several combinations, synonyms and related search terms to “Spermatocytic tumor” or “Spermatocytic seminoma”. To capture all relevant literature, no interventions, controls or specific outcomes were predefined in the search strategy. Non-English literature was excluded unless the abstract was available in English or the full text in French, Spanish, Italian or German. Additionally, the reference lists of the identified publications were screened manually to identify further studies. The detailed search strategy is shown in Appendix 1.
Duplicate articles were filtered using the “close match function” of Endnote and manual de-duplication. Two authors (JG, KS) screened the titles and abstracts independently to select publications that fulfilled the eligibility criteria and came to a consensus about the inclusion of those studies. Data of the same study that appeared in multiple publications were counted only once in the synthesis. Disagreements were discussed and resolved by consensus or by third-party arbitration (CDF).
Types of studies and participants
We included any case reports, clinical case series and other reports describing patients with testicular spermatocytic tumors.
Types of outcome measures included
Studies reporting clinicopathological variables, treatments of local or metastatic disease, site of metastases, disease-free, cancer-specific or overall survival were eligible for this review.
Assessment of risk of bias
The quality of the included articles was assessed based on the Cochrane Tool of risk of bias and the ROBINS-I assessment tool for non-randomized studies. In case of disagreement, a consensus will be found through discussion. The evaluated domains included: performance bias, detection bias, attrition bias, incomplete outcome data and selective outcome reporting.
Data extraction
A data extraction sheet (based on the Cochrane Consumers and Communication Review Group’s data extraction template) was developed and adapted after pilot testing on ten randomly selected eligible studies. Data on study design, patient characteristics, clinicopathological risk factors, treatment and follow-up were collected. One investigator (KS) extracted the data, and a second one (JG) reviewed the extracted data. Disagreements were discussed and resolved by consensus or by third-party arbitration (CDF). Given the fact that only retrospective case series and reports were identified we refrained from a study-base meta-analysis but whenever feasible, data were extracted on an individual patient level.
Statistical analysis
Descriptive data are presented as median, interquartile range (IQR) and range. To estimate medians using medians of cohort studies and individual patient age of single case presentation we used weighted medians. The results for continuous normally distributed variables are expressed as mean ± standard deviation (SD) and compared using Student’s t tests. Continuous non-normally distributed variables are presented as median and interquartile ranges (IQR) and compared using Wilcoxon rank-sum tests. The results for categorical variables are presented as percentage. A p value of < 0.05 was considered significant. All statistical tests were two-sided.
All statistical tests were two-sided. All p values < 0.05 were considered statistically significant and all analyses are considered exploratory and hypothesis generating.
Results
Studies
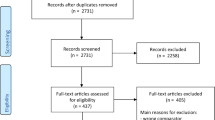
After deduplication, 3542 publications met the initial search criteria of which 62 publications were eligible for full text review after title and abstract had been checked. We screened the full text of 62 manuscripts and finally included 56 studies and 146 patients (Fig. 1, Supplementary Table 1). There were no randomized controlled or prospective trials and all includes case reports and series were rated as low quality of evidence.

Flow chart of the study selection process
Demographics, clinical symptoms and laboratory findings
Median patient age at diagnosis was 56.5 (range 25–94) years. The vast majority presented with testicular pain and/or enlargement. In a few cases, the diagnosis was confirmed after the work-up of other symptoms including infertility (3 patients), metastases (2 patients), hydrocele (2 patients), back pain (1 patient) or weight loss (1 patient). No case had an association with either changes in testosterone, estrogen levels or clinical signs of gynecomastia. Sonography details were infrequently reported like calcification in one and heterogeneous appearance in another patient. Serum tumor markers of germ cell cancers including alpha-fetoprotein (AFP), beta-human chorionic gonadotropin (bHCG), and lactate dehydrogenase (LDH) were reported in 46, 47 and 25 patients and elevated in 1, 2, and 3 patients, respectively.
Local treatment and pathological findings
The majority of orchiectomies was performed without frozen section analyses (96%). Finally, all patients underwent orchiectomy and no single case testis-sparing surgery (TSS). One patient with metastatic disease deteriorated quickly before orchiectomy was performed. The median tumor mass size was 70 mm (IQR 50–95, range 11–280). The most frequently described pathological findings included high mitotic activity in 30 patients, angiolymphatic invasion in 25 patients, CD117 in 25 patients, intratubular growth in 21 patients, necrosis in 20 patients, pleomorphism in four patients whereas other findings such as margins status, extracapsular extension, spermatic cord invasion, atypias, calcifications, p53, Ki67, Vimentin, Sall4 expression were reported in three patients or less. Histological variants included sarcoma or anaplastic subtypes and were present in 15 and 7 patients, respectively.
Adjuvant therapies
Six patients with localised disease were treated with adjuvant therapies. The first patient was treated with local and abdominal radiotherapy and chemotherapy (with vincristine, actinomycin D and cyclophosphamide) and remained disease-free at 2 months (Narang et al. 2012). The second patient was treated with adjuvant cisplatin, vinblastine and bleomycin but died because of a septic shock during chemotherapy (Musa et al. 1998). The third patient received two cycles of carboplatin but recurred 10 months after chemotherapy (Steiner et al. 2006). The fourth patient also received two cycles of carboplatin but remained disease-free after a follow-up of 8 months (Dundr et al. 2007). The fifth patient was treated with adjuvant radiotherapy to the pelvis and subclavicular region (30 Gray) (Caty et al. 1997). This patient died 1 year later because of a gastric lymphoma. The sixth patient was treated with adjuvant “20 sessions of radiotherapy” and no disease recurrence was noted (True et al. 1988).
Metastatic spread at initial stating and disease recurrence during follow-up
Any metastatic disease at any time point was reported in 14/146 (10%, 95% CI 5–15%) patients. Initial and follow-up staging was reported in 107/146 patients. At initial staging, metastases were present in 8/107 (7%) patients of which six had several sites of metastases [retroperitoneal lymph nodes (RPLN) (5), lung (2), liver (2), bone (1) and brain (1)] (Fig. 2). Data regarding follow-up were available in 89 patients. Median follow-up time was 44 months (IQR 12–96, range 1–324). During follow-up, one patient developed local recurrence and one patient a metachronous contralateral tumor. Metastatic relapse in patients with localised disease at diagnosis was reported in 6/89 (7%) at the following locations RPLN (4), lung (4), liver (1) and brain (1). Metastases during follow-up were diagnosed after a median follow-up of 5.5 months (range 2–21 months).

Anatomical locations of metastases in 107 patients of which staging was reported and 89 with follow-up examinations. Whereas 8/107 patients had metastatic disease at initial staging, 6/89 patients developed metastatic disease during follow-up
Risk factors and treatment of metastatic disease
Patients with sarcoma or anaplastic subtypes showed metastatic disease in 47% and 29%, respectively (Table 1). Compared to pure spermatocytic tumors, patients with histological variants (sarcoma or anaplastic subtype) were more likely to have metastatic disease (6/124 (5%) vs 9/22 (41%), p < 0.001) (Table 2). Patients with metastatic disease had larger primary tumors (92.5 vs 67.5 mm, p = 0.05). Survival of patients with metastatic disease ranged from 1 to 25 months. Radiotherapy or systemic therapy showed only partial responses in a few patients (Table 3). One patient with retroperitoneal relapse 10 months after orchiectomy and two cycles of adjuvant chemotherapy underwent laparoscopic RPLN-dissection. The specimen revealed spermatocytic tumor histology and adjuvant BEP was administered and the patient remained free of disease 3 years after chemotherapy (Steiner et al. 2006).
Risk of bias assessment
Our risk of bias assessment using the Cochrane tool of risk of bias showed high risk of bias for the included records due to fact that all available literature on the subject exclusively consists of retrospective case reports and small case series. Sources of bias included publication bias for cases with rare histologic variants or metastatic disease, lack of a priori protocols, missing data and inconsistency in outcome reporting (Supplementary Table 2). Predefined protocols (PRIMSA and Prospero) were used to identify and deter a possible reporting bias by determining our research question beforehand.
Discussion
This systematic review summarizes the available literature regarding local and systemic management, staging and follow-up for patients with testicular spermatocytic tumors.
However, as spermatocytic tumors of the testis are rare, the published literature consists only of retrospective studies, so that this review is prone to bias. Because spectacular and rare clinical findings are more likely to be published, this literature synthesis and especially the high incidence of metastastic disease of 7% is likely positively affected by publication bias. For example, Hu et al. described sarcoma subtypes in only 2 out of 83 spermatocytic tumors (2%), whereas our review identified 22 out of 146 (15%) thus pointing to a selection bias of this review. Furthermore, staging was not reported in all cases and may further influence the estimated proportions regarding metastatic disease and relapse. Our search criteria were designed and reviewed both by clinicians and librarians and were predefined in a peer-reviewed protocol. However, it is possible that not all potentially relevant studies were identified because of undetected imprecision of our search strategy, which would have to be classified as another potential source of bias. In the absence of larger and/or prospective studies, the current analysis is unique and may help physicians to select the most appropriate treatment modality for patients with spermatocytic tumors of the testis. Until more evidence becomes available, our results represent a basis for informed decision making in everyday clinical practice. Due to the absence of available retrospective or ongoing prospective trials, we recently opened the OrphAn Testis Histologies (OATH) Registry and encourage collaborators to contribute data of patients with rare testis cancer histologies (http://bit.ly/OATH-registry).
Despite all limitations, we believe that our results may facilitate decision making in a disease in which no clinical trials will be conducted in the near future and several observations can be made and clinical implications be suggested. First, patients with testicular spermatocytic tumors have a median age of 56 years, a large median tumor size of 70 mm, present nearly exclusively with local symptoms of testicular tumors and tumor markers of germ cell tumors are hardly ever elevated. Second, although spermatocytic tumors are by definition not associated with GCNIS, the large average tumor size and absence of limited with testis-sparing surgery, orchiectomy represents the gold standard. Orchiectomy instead of testis sparing surgery should also be standard of care because in our experience of the difficulty to differentiate spermatocytic tumors from pure seminoma in frozen section analysis. Third, we listed for the first time the most common sites of metastatic disease, which may guide staging modalities. Patients should be staged with computerized tomography (CT) including the chest, abdomen and pelvis as metastases at initial staging or at relapses have been described in these locations. Based on our findings that all patients with recurring metastatic aggressive disease were diagnosed within the first years, follow-up with CT could probably be limited to the first years. Fourth, the role of adjuvant chemotherapy and radiotherapy remains ill-defined and cannot be recommended. Fifth, surgical resections and chemotherapy was the only treatment sequence that was able to induce complete response in one metastatic patient whereas chemotherapy alone did not lead to complete response. Therefore, in contrast to germ cell cancers, spermatocytic tumors may represent a chemotherapy resistant entity which may also be responsible for the very poor prognosis of patients with metastatic disease of only a few months to years. Overall, the level of evidence to recommend any therapy option is minimal and recommendations rely mainly on expert knowledge. Therefore, we encourage clinicians to discuss patients with metastatic disease with an expert center including review of histopathology and imaging and to discuss either aggressive surgical approaches and/or chemotherapy.
Conclusion
The available literature provides only scarce evidence regarding the optimal management of patients with spermatocytic tumors. This so far largest retrospective collection provides several recommendations based on a low level of evidence and let us conclude that spermatocytic tumors are not only a biologically but also clinically distinct entity, should not be managed like germ cell cancers and require thorough multidisciplinary evaluation, especially in advanced stages. To further expand those recommendations, we set-up a multicenter registry and invite other centers to contribute their experiences in patients with spermatocytic tumors.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Albores-Saavedra J, Huffman H, Alvarado-Cabrero I, Ayala AG (1996) Anaplastic variant of spermatocytic seminoma. Hum Pathol 27:650–655
Bohm E, Walz PH, Tan KH (1993) A combination of spermatocytic and classic seminoma, mature teratoma and carcinoma in situ of the testis. An attempt at an etiologic explanation. Zentralblatt fur Pathologie 139:255–260
Caty A, Bertrand P, Castelain B, Mazeman É (1997) Spermatocytic seminoma. Report on 4 cases. Bull Cancer 84:212–214
Chelly I, Mekni A, Gargouri MM et al (2006) Spermatocytic seminoma with rhabdomyosarcomatous contingent. Progrès en urologie 16:218–220
Choi SK, Kim T, Yoo KH (2017) Spermatocytic seminoma with brain metastasis. Pathol Oncol Res 23:223–224
Dundr P, Pesl M, Povysil C, Prokopova P, Pavlik I, Soukup V, Dvoracek J (2007) Anaplastic variant of spermatocytic seminoma. Pathol Res Pract 203:621–624
Floyd C, Ayala AG, Logothetis CJ, Silva EG (1988) Spermatocytic seminoma with associated sarcoma of the testis. Cancer 61:409–414
Gentile G et al (2014) First case of bilateral, synchronous anaplastic variant of spermatocytic seminoma treated with radical orchifunicolectomy as single approach: case report and review of the literature. Archivio italiano di urologia, andrologia : organo ufficiale [di] Societa italiana di ecografia urologica e nefrologica 86:41–42. https://doi.org/10.4081/aiua.2014.1.41
Looijenga LH et al (2006) Genomic and expression profiling of human spermatocytic seminomas: primary spermatocyte as tumorigenic precursor and DMRT1 as candidate chromosome 9 gene. Cancer Res 66:290–302. https://doi.org/10.1158/0008-5472.can-05-2936
Matoska J, Talerman A (1990) Spermatocytic seminoma associated with rhabdomyosarcoma. Am J Clin Pathol 94:89–95
Matoška J, Ondruš D, Horňák M (1988) Metastatic spermatocytic seminoma. A case report with light microscopic, ultrastructural, and immunohistochemical findings. Cancer 62:1197–1201
Menon S, Karpate A, Desai S (2009) Spermatocytic seminoma with rhabdomyosarcomatous differentiation: a case report with a review of the literature. J Cancer Res Ther 5:213–215. https://doi.org/10.4103/0973-1482.57131
Mikuz G, Böhm GW, Behrend M et al (2014) Therapy-resistant metastasizing anaplastic spermatocytic seminoma: a cytogenetic hybrid. Anal Quant Cytol Histol 36:177–182
Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM (2016) The 2016 WHO classification of tumours of the urinary system and male genital organs-part A: renal, penile, and testicular tumours. Eur Urol 70:93–105. https://doi.org/10.1016/j.eururo.2016.02.029
Moher D, Liberati A, Tetzlaff J, Altman DG (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol 62:1006–1012. https://doi.org/10.1016/j.jclinepi.2009.06.005
Moher D et al (2015) Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev 4:1
Musa FM, Kuwata Y, Zhang XH, Takenaka I (1998) Spermatocytic seminoma: a case report. Nishinihon J Urol 60:558–560
Narang V, Gupta K, Gupta A, Kumar S (2012) Rhabdomyosarcomatous differentiation in a spermatocytic seminoma with review of literature. IJU 28:430–433. https://doi.org/10.4103/0970-1591.105760
Robinson A, Bainbridge T, Kollmannsberger C (2007) A spermatocytic seminoma with rhabdomyosarcoma transformation and extensive metastases. Am J Clin Oncol 30:440–441
Rosenberg C et al (1998) Chromosomal constitution of human spermatocytic seminomas: comparative genomic hybridization supported by conventional and interphase cytogenetics. Genes Chromosomes Cancer 23:286–291
Schoborg TW, Whittaker J, Lewis CW (1980) Metastatic spermatocytic seminoma. J Urol 124:739–741
Steiner H, Gozzi C, Verdorfer I, Mikuz G, Bartsch G, Hobisch A (2006) Metastatic spermatocytic seminoma: an extremely rare disease. Eur Urol 49:183–186. https://doi.org/10.1016/j.eururo.2005.08.020
Stueck AE, Grantmyre JE, Wood LA, Wang C, Merrimen J (2017) Spermatocytic tumor with sarcoma: a rare testicular neoplasm. Int J Surg Pathol 25:559–562. https://doi.org/10.1177/1066896917706156
Trivedi P, Pasricha S, Gupta A (2011) Spermatocytic seminoma associated with undifferentiated sarcoma: a rare case report. Indian J Pathol Microbiol 54:138–140
True LD, Otis CN, Delprado W, Scully RE, Rosai J (1988) Spermatocytic seminoma of testis with sarcomatous transformation: a report of five cases. Am J Surg Pathol 12:75–82
Wetherell D, Lawrentschuk N, Gyomber D (2013) Spermatocytic seminoma with sarcoma: an indication for adjuvant chemotherapy in localized disease. Korean J Urol 54:884–887. https://doi.org/10.4111/kju.2013.54.12.884
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Grogg, J.B., Schneider, K., Bode, PK. et al. A systematic review of treatment outcomes in localised and metastatic spermatocytic tumors of the testis. J Cancer Res Clin Oncol 145, 3037–3045 (2019). https://doi.org/10.1007/s00432-019-03056-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-019-03056-1