
Abstract
Background
Dysregulation of the complement system has been shown to play a major role in the pathogenesis of age-related macular degeneration (AMD).
Methods
The current evidence from human studies derives from immunohistochemical and proteomic studies in donor eyes, genetic association studies, and studies of blood complement protein levels. These lines of evidence are corroborated by in vitro and animal studies.
Results
In AMD donor eyes, detection of complement proteins in drusen suggested local inflammatory processes involving the complement system. Moreover, higher levels of complement proteins in the Bruch’s membrane/choroid complex could be detected in AMD donor eyes compared to controls. A large number of independent genetic studies have consistently confirmed the association of AMD with risk or protective variants in genes coding for complement proteins, including complement factor H (CFH), CFH-related proteins 1 and 3, factor B/C2, C3 and factor I. Another set of independent studies detected increased levels of complement activation products in plasma of AMD patients, suggesting that AMD may be a systemic disease and the macula a vulnerable anatomic site of minimal resistance to complement activation. Genotype–phenotype correlations, including the impact of genetic variants on disease progression, gene–environment and pharmacogenetic interactions, have been investigated. There is evidence that complement gene variants may be associated with the progression from early to late forms of AMD, whereas they do not appear to play a significant role when late atrophic AMD has already developed. There are indications for an interaction between genetic variants and supplementation and dietary factors. Also, there is some evidence that variants in the CFH gene influence treatment effects in patients with neovascular AMD.
Conclusions
Such data suggest that the complement system may have a significant role for developing new prophylactic and therapeutic interventions in AMD. In fact, several compounds acting on the complement pathway are currently in clinical trials. Therapeutics that modulate the complement system need to balance inhibition with preservation of sufficient functional activity in order to maintain adequate immune responses and tissue homeostasis. Specifically, targeting the dysfunction appears more adequate than a global suppression of complement activation in chronic diseases such as AMD.
Similar content being viewed by others


Age-related macular degeneration (AMD), the leading cause of blindness in western societies, is a complex disease with genetic, environmental and demographic risk factors [1–4]. In recent years, there has been growing evidence that inflammatory processes, including dysregulation of the complement system, play a major role in the pathogenesis of AMD. The discovery of genetic polymorphisms in genes coding for complement proteins that affect patients’ susceptibility to AMD propelled research into establishing the complement system as a key component in the pathogenesis of AMD [1–5].
However, translation from the bench to the bedside is a complex process, and our understanding of the role of the complement system in AMD is still at a distance from the bedside. This review aims to discuss the significance of the complement system for the pathogenesis of AMD, and the resulting implications for current and future clinical application.
First, a brief overview of the complement system is provided, along with evidence of its role in AMD pathogenesis. Second, the clinical relevance of polymorphisms of the complement genes is addressed. This includes associations of the genetic variants with alterations of complement activation or with certain AMD subtypes, as well as their possible influence on AMD progression, response to treatment (pharmacogenetics), or interference with known modifiable risk factors for AMD (gene-environment interaction). Third, we summarize approaches that might provide us with future treatment options.
The complement system and its role in AMD pathogenesis
Based on specificity and immediacy, immune processes have been discerned into two main effector systems. Both, the adaptive and the innate immune system are tightly interconnected. The adaptive immune system is built around B- and T-lymphocytes which allow antigen-specific recognition and immunological memory. In contrast, the innate immune system acts less specifically but more instantaneously. One of its main effectors is the complement system, which encompasses more than 30 plasma proteins with a combined concentration of more than 3 g/l [6, 7]. The evolutionary preservation of this ancient immune system is a reflection of its biological importance for the integrity of the organism [6].
The complement system can be activated via three different pathways: the classic, lectin or alternative pathway. In each, a proteolytic cascade is amplified and eventually leads to the activation of a central protein, C3. Downstream from C3 activation, the effectors of the complement system may be classified as anaphylatoxins (C3a, C5a; leading to inflammation), the membrane attack complex (MAC; leading to cell lysis), and opsonins (leading to opsonization).
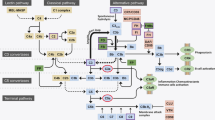
The classic pathway is activated when the complement protein C1q binds to antibodies attached to antigen. In contrast, the lectin pathway uses pattern recognition receptors (PRRs) such as mannose-binding lectin (MBL) to recognize pathogen-associated molecular patterns (PAMPs). The alternative pathway of complement (APC) will be described in greater detail, as current evidence suggests that it is the most important in relation to the pathogenesis of AMD. For more in-depth information on the complement system, the reader is referred to excellent reviews [6, 7].
The APC (Fig. 1) is initially activated by a constant low-level spontaneous hydrolysis of C3. Binding of C3 to factor B allows factor D to cleave factor B into Ba and Bb. One of the resulting products, C3bBb, is a C3 convertase that initiates an amplification loop, producing more C3b and C3a from C3. Uncontrolled activation of the APC would lead to self-tissue damage. Therefore, regulators are necessary to prevent the inappropriate activation and action of the APC [6]. Such regulators may accelerate the decay of preformed C3 convertases, e.g., decay-accelerating factor (DAF; CD55), complement receptor 1 (CR1), complement factor H (CFH), or prevent convertases to form by degrading their constituents such as C3b [6]. The latter is a function of factor I, which requires other complement regulators (CR1, CFH, membrane cofactor protein [MCP; CD46]) as cofactors. Both processes ultimately limit further complement activation. Other regulators may inhibit MAC-mediated cell lysis (CD59, vitronectin, S protein) or may cleave anaphylatoxins (carboxypeptidase N, B and R). The regulators of complement may further be classified as cell-bound (e.g., MCP, CR1, DAF, CD59) or as being located in the fluid phase (CFH, vitronectin, S protein). CFH is the most important regulator of the latter group.

Proteins of the alternative complement cascade are typed in black (see text). In red font are therapeutic agents currently in clinical trials. For further explanation of the therapeutic agents, see main text
Evidence for involvement of the complement system in AMD pathogenesis
There are currently three separate lines of evidence from human studies that support the involvement of the complement system in AMD pathogenesis (Fig. 2):
-
Immunohistochemical and proteomic studies in donor eyes
-
Genetic association studies
-
Studies of complement protein levels in peripheral blood

Three lines of evidence from human studies supporting involvement of the complement system in the pathogenesis of age-related macular degeneration. An increased local (demonstrated by immunohistochemical) and systemic (demonstrated by plasma protein studies) complement activation may be due to a genetic risk profile, and is possibly affected by various additional factors
The first line of evidence derives from immunohistochemical detection of proteins of the complement cascade, its regulators and other inflammatory markers. These were increased in donor eyes from AMD patients compared to controls and characteristically localized in drusen, the hallmark clinical finding of early AMD [5, 8–13]. Further supporting evidence came from a quantitative proteomics analysis of the macular Bruch’s membrane/choroid complex. In AMD donor eyes, many complement proteins were elevated compared to control eyes [14]. Complement proteins such as CFH are also present in normal eyes [15], and may therefore have a physiological function. The disease-associated variant of CFH was shown to have a lower affinity to Bruch’s membrane [16], which might lead to an uncontrolled and thus increased local activation of the alternative complement cascade.
Evidence for a genetic component of AMD susceptibility arises from twin and family studies that have consistently shown increased susceptibility in individuals with positive family history [17–23]. In 2005, genetic association studies revealed significant associations of polymorphisms in the complement factor H (CFH) gene with an increased or decreased risk for AMD [15, 24–26]. The significance of the complement system was further substantiated by the identification of additional genes coding for proteins of the complement system and their association with an increased or decreased risk to develop AMD. These include genes coding for complement factor B/C2 (CFB) [27–29], C3 [30–32], factor I (FI) [33, 34], and CFH-related proteins 1 and 3 [35, 36]. The associations between these variants and AMD, however, appear to be substantially weaker than for variants in CFH. A large number of independent studies have since consistently confirmed the association of AMD with risk-modifying complement gene polymorphisms. For a more in-depth discussion of genetic AMD susceptibility, the reader is referred to other recent thorough reviews [1, 37–40].
The third line of evidence derives from studies of blood complement protein levels. The data show that AMD patients have increased systemic complement activation as measured in peripheral blood (see next section) [41–43].
The evidence from human studies is supported by a large number of in vitro and animal studies. Notably, subsequent functional in vitro analysis provided evidence for a biological relevance of CFH variants. The altered protein structure of the CFH Y402H “at risk” variant results in a decreased binding affinity to target molecules such as C-reactive protein and heparin to cell surfaces and Bruch’s membrane [16, 44–50]. In contrast, the protective CFH V62I variant has been found to be a stronger inhibitor of C3 convertase formation [51]. Furthermore, it has been shown that constituents of lipofuscin, the accumulation of which is part of the disease process in AMD, may activate complement [52]. Also, smoking as well as a low-grade immunoresponse against carboxyethylpyrrole (CEP) adducts that accumulate in the subretinal space may result in complement activation at the ocular fundus in mouse models [53, 54]. The transcriptional profiles of the RPE/choroid complex in aged (compared to young) mice showed a marked increase in proteins of the complement pathway [55]. Thus, major risk-conferring factors for AMD development, such as oxidative stress, ageing and smoking, have been linked to an activation of the complement system. Moreover, the RPE physiology is affected by complement activation, and the RPE is also capable of modulating its own complement protein production, suggesting that RPE cells may play an important role in regulating complement activation in the retina [56–59]. A CFH-/- mouse model has been described which develops a retinal phenotype in old age [60].
Clinical relevance of variations of complement genes
Systemic complement activation in AMD patients
Immunohistochemical studies on AMD donor eyes and data from genetic studies suggest that AMD pathogenesis involves mainly the APC. This led to the hypothesis that the relevant genetic polymorphisms, if biologically meaningful for AMD pathogenesis, would result in measurable differences in the activation status of the complement cascade. Indeed, the first study that comprehensively assessed plasma concentrations of APC proteins in AMD patients and controls found higher levels of activation products in the AMD cohort [41]. Specifically, all tested activation products (Ba, C3d, MAC, C3a, C5a), especially the markers of chronic complement activation (Ba and C3d; p < 0.001), were significantly elevated. Similar alterations were observed in the activating regulator factor D, but not in C3, C4 or CFH. The increased concentration of protein markers of the APC correlated with CFH haplotypes in patients and controls, suggesting a genetically controlled activation of the APC. A subsequent study in a larger independent cohort of patients and controls essentially confirmed these results, showing that both plasma protein levels and genetic markers were individually predictive of having AMD [43]. A one standard deviation change in levels of complement substrate (factor B), regulator (factor D), and activation products (Ba and C3d) was associated with an approximately 5-fold increase in AMD risk (Fig. 3).

Prediction of AMD and impact of plasma complement levels on AMD risk. Shown are the odds ratios for each combination of a one standard deviation change in plasma levels of factor D, factor B, C3d, and Ba on the risk to develop AMD relative to a reference group (R) having mean levels of all four proteins. Numbers on the x and y axes represent standard deviation changes above (1), changes below (−1), and at the mean (0) of the corrected and standardized plasma levels for each protein. From Hecker et al. [43]; by permission of Oxford University Press
Support for these observations comes from biochemical in vitro studies already mentioned, which may explain the variable concentration of activation products downstream from CFH due to a difference in biological activity related to the CFH variant. The study by Hecker et al. furthermore revealed an association of complement activation with genetic polymorphisms in CFB, and suggested that activation of the APC in blood increases with age [43]. A trend was observed for greater increases in plasma protein levels of factor D, factor B, Ba and C3d in advanced subtypes of AMD, suggesting that complement activation in the blood could be associated with progression of AMD. A third study focussing on patients with advanced AMD also found an increased complement activation in AMD patients: studying a slightly different set of complement proteins, Reynolds and co-workers found an increased plasma concentration of C5a and Bb, independent of genotype. The group had also found decreased CFH plasma concentration in patients with geographic atrophy, a finding not reported by the two other studies. Furthermore, the study provided evidence for an association of increased body mass index with complement activation fragments [42].
Further support for the suggested association between abnormal complement activation and AMD came from a large case control study that revealed an association of AMD with diseases known to be associated with systemic complement activation [61]. AMD patients were found to have systemic lupus erythematosus (confounder adjusted OR = 1.83) and glomerulonephritis (adjusted OR = 1.46) more often than controls. Moreover, patients with membranoproliferative glomerulonephritis (MPGN) type 2, a disease with uncontrolled complement activation [62], may show a phenocopy of the retinal findings, such as drusen, that are usually observed in patients with AMD [63–66].
Notably, these studies provide a rationale for future clinical trials that aim at a systemic modulation of complement activation in order to prevent AMD. However, evidence available so far relates to the link between complement activation and the risk to develop the disease. Currently, there is no evidence from human studies that complement activation is still pivotal when late forms of AMD have already developed.
Complement gene variants and AMD subtypes
To date, none of the associated complement gene variants have shown a clear pattern of preference for the development of either choroidal neovascularisation (CNV) or geographic atrophy [67–71]. This may suggest that complement variants are equally important for the development of both forms of late AMD, and additional genetic and/or environmental factors may be required to determine if either geographic atrophy or CNV develops. It remains conceivable that all previous studies have not been sufficiently powered in order to detect an effect, in that most studies are biased towards CNV patients. Further studies have analyzed a potential correlation between the CFH-risk allele Y402H and specific subtypes of neovascular AMD [72–75]. Due to the heterogeneous results in those studies, clear conclusions with regard to a preferential occurrence of classic or occult neovascular membranes can currently not be drawn. Possibly, severity and age of onset of neovascular membranes in AMD patients may be influenced by the genetic background, as suggested by Leveziel et al. [76]. Shuler et al. analyzed phenotypic characteristics in a cohort of 956 AMD patients, and identified only peripheral reticular pigmentary change as a phenotypic feature associated with the common Y402H risk variant [71].
The phenotype of basal laminar drusen (“cuticular drusen”) is similar to, yet distinct from AMD. Basal laminar drusen have also been shown to be associated with CFH variants [66]. The Y402H variant may be present in up to 70% of patients with basal laminar drusen [77], which is higher than usually observed in populations with typical AMD. Early-onset basal laminar drusen were reported to be associated with heterozygous nonsense, missense or splice-variants of CFH in combination with the Y402H variant [78].
Polypoidal choroidal vasculopathy (PCV) has been described as a separate clinical entity differing from neovascular AMD and other diseases associated with subretinal neovascularization [79], and it remains controversial as to whether or not PCV represents a sub-type of neovascular AMD [80]. Patients with PCV tend to be younger, the disease is more prevalent in Oriental races, and eyes with PCV lack drusen as a characteristic sign of early AMD. In a comprehensive examination of the CFH gene, Kondo and co-workers found a strong association with the I62V variant in a cohort of 130 Japanese PCV patients [81]. Notably, they found no significant association of the Y402H variant with PCV, which contrasts with its marked effect on AMD susceptibility in Caucasians. Similar conclusions were derived from a study by Lee et al. in a Chinese population [82]. However, a recent and better powered study (408 patients with typical AMD, 518 patients with PCV, 1,351 control samples) also identified an association of PCV with the Y402H allele in addition to the association with the I62V variant [83]. Gotoh et al. found no difference in incidence of the CFH Y402H genotype between patients with exudative AMD and PCV, and a recent study on a Caucasian population with PCV and AMD also suggested that both diseases are genetically similar on the CFH and CFB/C2 locus [84]. The latter study included the Y402H but not the I62V variant. These genetic studies provide evidence for a similar pathogenetic background of the two phenotypically similar diseases, namely an involvement of the complement system. Different polymorphisms may have different functional consequences on the protein level, and might at least in part account for the different phenotype. The eventual disease phenotype may furthermore be determined by additional genetic and/or environmental factors. Only few data are available on the association of retinal angiomatous proliferation (RAP) with polymorphisms in genes coding for complement proteins. In a Japanese population, a correlation with RAP was found for the CFH I62V variant but not for the Y402H variant [83], while in an Austrian population, an association of RAP was found for the Y402H variant [72].
Complement gene variants and progression of AMD
In two independent cohorts, Magnusson and co-workers showed that the Y402H variant conferred a similar risk of early and late AMD [68]. Also, Farwick and colleagues reported the CFH variants to be significantly related to the development of early but not with progression to late AMD [85]. This suggested that additional factors may be required to explain disease progression. However, data from the population-based Rotterdam study revealed an increasing odds ratio with more progressed disease stages [86], and two further studies found an increased risk to progress from early to later disease stages in patients with the CFH [87–89] and C3 [89, 90] risk genotypes. Results for the C2/CFB locus were inconsistent, possibly due to different single nucleotide polymorphisms (SNPs) tested [89, 90].
It should be noted, however, that none of these studies has addressed the question of whether genetic variants are associated with disease progression once late AMD has developed. Nevertheless, it is the progression of the two late forms, geographic atrophy and choroidal neovascularisation, which are probably the most important with regard future therapeutic intervention. This issue was recently addressed in a longitudinal association study investigating variants in CFH, C3 and ARMS2, and progression of geographic atrophy in a large cohort of AMD patients with pure bilateral geographic atrophy [91]. It was found that all genetic risk variants were strongly associated with the risk of developing geographic atrophy, whereas there was no association with disease progression once geographic atrophy had already developed. It is suggestive that other susceptibility factors may influence disease progression in geographic atrophy [91]. This was very recently confirmed in a study of Klein et al. where — at least for variants in the complement genes — there was no association with the progression of geographic atrophy [92].
Gene-environment interaction: nutrition, supplementation and smoking
The only protective factor for AMD known to date are antioxidants. The randomized clinical Age-Related Eye Disease Study (AREDS) showed that a combination of zinc, β-carotene, vitamin C, and vitamin E reduced the risk of progression from intermediate to advanced AMD by 25% [93]. There is also preliminary evidence of a protective effect of omega-3 fatty acids [94–96].
Data on the interaction between nutrition or supplementation and genetic variants in regard to the risk to develop AMD has been limited, but recently two large studies, the Age-Related Eye Disease Study (AREDS), being clinic-based, and the Rotterdam study, being population-based, provided evidence for an interaction.
In the AREDS, Klein and co-workers studied a subset of 876 participants who were considered at high risk of progressing to advanced AMD [97]. They found a treatment interaction of the CFH Y402H polymorphism with the AREDS medication. Interestingly, supplementation resulted in a 68% reduction in the rate of progression in the subgroup with the homozygous non-risk genotype, compared to a reduction of only 11% in the subgroup with the homozygous risk genotype. Thus, the data suggest reduced benefit of AREDS supplementation for patients with the CFH Y402H risk genotype. Further sub-analysis found the genotype–treatment interaction to be explained by the zinc component, since an interaction was observed in the groups taking zinc versus those taking no zinc, but not for groups taking antioxidants compared with groups taking no antioxidants. No significant CFH genotype–treatment interaction effects on progression were observed when the analysis additionally included patients with an earlier disease stage (AREDS category 2) [87].
Conflicting results were recently reported by Ho et al. investigating 2,167 individuals from the Rotterdam Study at risk of AMD [98]. They assessed biological interaction with genetic variants by calculating the synergy indices. In a mean follow-up period of 8.6 years, 517 participants developed early AMD. Significance of the synergy index supported the possibility of biological interaction between CFH Y402H and zinc, β-carotene, lutein/zeaxanthin, and eicosapentanoic/docosahexaenoic acid (EPA/DHA). Homozygotes for CFH Y402H with dietary intake of zinc in the highest tertile reduced their hazard ratio of developing early AMD from 2.25 to 1.27. For intakes of β-carotene, lutein/zeaxanthin, and EPA/DHA, these risk reductions were from 2.54 to 1.47, 2.63 to 1.72, and 1.97 to 1.30, respectively [98].
The discrepancies between the two studies may be explained by the different design: AREDS is clinic-based, and thus the study population is affected by early or late AMD at baseline, whereas the cohort of the Rotterdam study consisted of participants without any sign of AMD. Consequently, the outcome event was different: for AREDS it was the progression from early to late AMD or from unilateral to bilateral late AMD, whereas in the study of Ho et al. it was incident early AMD.
Smoking is by far the strongest environmental risk factor of AMD susceptibility. Smoking increased the risk of AMD additive to the genetic predisposition due to variants in the CFH gene [86, 99–101]. Smoking as well as an increased body mass index (BMI) were independently related to advanced AMD, controlling for the genotype [70]. Smoking and having the CFH 402H variant independently increase risk of neovascular AMD [102, 103]. Smoking increased the odds of disease progression due to the CFH [87, 89] and other genetic risk variants [89]. Smoking was independently related to AMD, with a multiplicative joint effect with genotype on AMD risk [89]. Therefore, there appears to be no interaction between smoking and CFH genotypes.
Variations of complement genes and response to treatment: pharmacogenetics
Intravitreal injections of vascular endothelial growth factor (VEGF)-A inhibitors have recently revolutionized the therapy of neovascular AMD [104]. There are, however, surprisingly few data on the interaction of treatment effect and genetic variants. A retrospective analysis of 86 patients treated with intravitreal bevacizumab revealed a significantly worse visual acuity outcome in patients with the CFH Y402H risk genotype (CC) compared with those with the TC and TT genotypes [105]. The same researchers retrospectively investigated the pharmacogenetic interaction between CFH variants and the treatment effect of ranibizumab [106]. In their cohort of 156 patients, there was no effect for the primary outcome measure, visual acuity. However, the data suggested that patients homozygous for the Y402H risk allele may have a higher risk of requiring more ranibizumab injections. The authors hypothesized that the higher inflammatory activity found in genetically predisposed patients could favor recurrence of neovascularization or reduce its response to anti-VEGF treatment. Obviously, further studies are needed to explore significant interaction between genetic variants and anti-VEGF-A treatment effects.
Several studies assessed a potential association between the CFH Y402H genotype and response to photodynamic therapy (PDT) [107–110]. The largest study included 273 patients treated with PDT and a median follow-up time of 19.8 months. There was no significant difference in genotype distribution between a PDT-positive and a PDT-negative response group (the latter being defined as visual acuity of <6/60 or loss of 3 lines of vision at final visit) [109]. Similar results were presented in a Finnish study with 88 participants [111]. There was also no significant difference in number of PDT treatments needed depending on genotypes. Two smaller studies with shorter follow-up times suggested either a worse [107] or a better [108] outcome (visual acuity) in the patient group homozygous for the CFH Y402H risk allele. A recent study from Japan found an association of CFH variants with the mean time interval from the initial treatment to the time of recurrence [110]. These overall conflicting results may simply be due to limitations in statistical power.
It is likely that pharmacogenetic studies will play a more important role when testing compounds that target and modulate the complement system. AMD patients with “at-risk” genotypes might be more responsive to such interventions and therefore, specific genetic markers will probably impact on a meaningful allocation of specific treatments. As an alternative to genetic testing, protein-based methods have now been developed that make it possible to distinguish CFH risk variants in plasma [112, 113].
Emerging pharmacological intervention
Since the link between complement activation and AMD susceptibility has unequivocally been established, modulation of the complement system appears to be a reasonable strategy for reducing the risk of developing AMD, for preventing progression from early to late forms of AMD, for treating late AMD, or for optimizing currently available treatments such as VEGF-A inhibition.
Several compounds are currently in phase 1 or 2 clinical trials (see Fig. 1; clinicaltrials database accessed August 4, 2010).
POT-4 (Potentia Pharmaceuticals/Alcon) is an analogue of the small cyclic synthetic peptide compstatin, an inhibitor of the central complement protein C3. A sustained release formulation aims at providing therapeutic drug concentrations for several months after intravitreal injection. A prospective, uncontrolled, non-randomized, dose-escalating, pilot phase 1 safety study in AMD patients with subfoveal CNV has been completed (ClinicalTrials.gov Identifier: NCT00473928).
Eculizumab (Soliris®, Alexion Pharmaceuticals) is a monoclonal antibody inhibiting C5. The drug is approved as systemic treatment for paroxysmal nocturnal hemoglobinuria (PNH), a disease characterized by the absence of CD59 expression of erythrocytes. Eculizumab is currently being investigated in a phase 2 trial (ClinicalTrials.gov Identifier: NCT00935883) in patients with non-exudative AMD (drusen or geographic atrophy). In the randomized, double-arm, double-masked study, patients receive the drug via an intravenous infusion. Primary outcome measures are the growth of geographic atrophy and the change in drusen volume, respectively.
ARC1905 (Ophthotech Corporation) is an anti-C5-aptamer that is injected intravitreally with currently two registered phase 1 clinical trials. ARC1905 is meant to bind C5 to prevent its interaction during activation of the complement cascade. In one clinical trial, ARC1905 is used for neovascular AMD in combination therapy with either multiple doses or with only one induction dose of intravitreal ranibizumab in a non-randomized, open-label, uncontrolled, safety study (ClinicalTrials.gov Identifier: NCT00709527). The second trial (ClinicalTrials.gov Identifier: NCT00950638), a randomized, open-label, dose comparison study, aims at elucidating the safety profile of intravitreal ARC1905 application in dry AMD (drusen and/or geographic atrophy).
FCFD4514S (Genentech / Roche) is an anti-complement factor D antibody fragment that is injected intravitreally. Similarly to ARC1905, its safety is currently in evaluation in a phase 1 trial in patients with dry AMD (geographic atrophy; ClinicalTrials.gov Identifier: NCT00973011).
Several other compounds are currently being tested in a pre-clinical phase that aims either at inhibiting the effect of activated complement proteins or at normalizing an increased activation of the complement cascade.
Conclusions
There is strong evidence from human immunohistochemical, genetic and proteomic studies supporting a major role of the complement system in AMD pathogenesis, and this is further supported by a large number of in vitro and animal studies. Genetically controlled systemic complement activation may have a major impact on the macula as a “locus minoris resistentiae”. Moreover, altered complement proteins may have specific local effects such as an impaired control of complement activation at the level of the RPE and Bruch’s membrane.
Therapeutics that aim at modulating processes of the complement cascades will need to balance the beneficial effects of inhibition with the preservation of sufficient functional activity for immune responses and tissue homeostasis [114]. This appears most important in chronic diseases such as AMD where long-term treatment appears essential to prevent vision loss due to conversion from early into late disease stages. Ideally, the pharmacological effect would act on the key dysregulated elements of AMD pathophysiology, leaving the remaining complement system largely unaffected.
There is still a lack of knowledge about the impact of individual elements of the complement system on AMD development, phenotypic variability and progression. It would be desirable to target mechanisms that are involved in disease progression and not merely in disease susceptibility. Moreover, there is as yet insufficient evidence to determine if dysregulation of either the systemic or the local complement components or both are the major contributors in AMD pathogenesis. Such knowledge would probably affect the preferred route of drug administration. A more detailed summary on potential and current strategies of therapeutic complement inhibition is provided elsewhere [114, 115].
Due to the rather low yearly progression rate from early to late AMD stages, large patient cohorts may have to be studied over many years to detect a prophylactic effect of any intervention on conversion rates to late disease stages. The AREDS has shown that early AMD patients with a high risk of progression must be identified to provide sufficient power to detect a prophylactic effect.
In emerging treatment trials, adequate disease monitoring will be essential. Compounds targeting the complement system might be most beneficial in early AMD stages where treatment effects are likely to be subtle. Biomarkers such as drusen volume have not been unequivocally qualified as meaningful surrogate endpoints. Thus, high resolution multi-modal functional and/or morphological assessment strategies may be needed in order to identify and correctly interpret possible treatment effects [116].
References
Ding X, Patel M, Chan CC (2009) Molecular pathology of age-related macular degeneration. Prog Retin Eye Res 28:1–18
Coleman HR, Chan CC, Ferris FL III, Chew EY (2008) Age-related macular degeneration. Lancet 372:1835–1845
Jager RD, Mieler WF, Miller JW (2008) Age-related macular degeneration. N Engl J Med 358:2606–2617
Swaroop A, Chew EY, Rickman CB, Abecasis GR (2009) Unraveling a multifactorial late-onset disease: from genetic susceptibility to disease mechanisms for age-related macular degeneration. Annu Rev Genomics Hum Genet 10:19–43
Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Bowes RC, Bok D, Hageman GS, Johnson LV (2009) The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res 29:95–112
Dunkelberger JR, Song WC (2010) Complement and its role in innate and adaptive immune responses. Cell Res 20:34–50
Walport MJ (2001) Complement. First of two parts. N Engl J Med 344:1058–1066
Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG (2002) Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA 99:14682–14687
Johnson LV, Leitner WP, Staples MK, Anderson DH (2001) Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res 73:887–896
Anderson DH, Mullins RF, Hageman GS, Johnson LV (2002) A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol 134:411–431
Mullins RF, Russell SR, Anderson DH, Hageman GS (2000) Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J 14:835–846
Hageman GS, Luthert PJ, Chong NHV, Johnson LV, Anderson DH, Mullins RF (2001) An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res 20:705–732
Mullins RF, Aptsiauri N, Hageman GS (2001) Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye 15:390–395
Yuan X, Gu X, Crabb JS, Yue X, Shadrach K, Hollyfield JG, Crabb JW (2010) Quantitative proteomics: comparison of the macular Bruch membrane/choroid complex from age-related macular degeneration and normal eyes. Mol Cell Proteomics 9:1031–1046
Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, Sangiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308:385–389
Clark SJ, Perveen R, Hakobyan S, Morgan BP, Sim RB, Bishop PN, Day AJ (2010) Impaired binding of the AMD-associated complement factor H 402H allotype to Bruch’s membrane in human retina. J Biol Chem 285:30192–30202
Seddon JM, Cote J, Page WF, Aggen SH, Neale MC (2005) The US twin study of age-related macular degeneration: relative roles of genetic and environmental influences. Arch Ophthalmol 123:321–327
Heiba IM, Elston RC, Klein BE, Klein R (1994) Sibling correlations and segregation analysis of age-related maculopathy: the Beaver Dam Eye Study. Genet Epidemiol 11:51–67
de Jong PT, Klaver CC, Wolfs RC, Assink JJ, Hofman A (1997) Familial aggregation of age-related maculopathy. Am J Ophthalmol 124:862–863
Klaver CC, Wolfs RC, Assink JJ, van Duijn CM, Hofman A, de Jong PT (1998) Genetic risk of age-related maculopathy. Population-based familial aggregation study. Arch Ophthalmol 116:1646–1651
Smith W, Mitchell P (1998) Family history and age-related maculopathy: the Blue Mountains Eye Study. Aust N Z J Ophthalmol 26:203–206
Luo L, Harmon J, Yang X, Chen H, Patel S, Mineau G, Yang Z, Constantine R, Buehler J, Kaminoh Y, Ma X, Wong TY, Zhang M, Zhang K (2008) Familial aggregation of age-related macular degeneration in the Utah population. Vision Res 48:494–500
Seddon JM, Ajani UA, Mitchell BD (1997) Familial aggregation of age-related maculopathy. Am J Ophthalmol 123:199–206
Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA (2005) Complement factor H variant increases the risk of age-related macular degeneration. Science 308:419–421
Edwards AO, Ritter R III, Abel KJ, Manning A, Panhuysen C, Farrer LA (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308:421–424
Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R (2005) A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA 102:7227–7232
Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Hageman GS, Dean M, Allikmets R, Chang S, Yannuzzi LA, Merriam JC, Barbazetto I, Lerner LE, Russell S, Hoballah J, Hageman J, Stockman H (2006) Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet 38:458–462
Jakobsdottir J, Conley YP, Weeks DE, Ferrell RE, Gorin MB (2008) C2 and CFB genes in age-related maculopathy and joint action with CFH and LOC387715 genes. PLoS One 3:e2199
Spencer KL, Hauser MA, Olson LM, Schmidt S, Scott WK, Gallins P, Agarwal A, Postel EA, Pericak-Vance MA, Haines JL (2007) Protective effect of complement factor B and complement component 2 variants in age-related macular degeneration. Hum Mol Genet 16:1986–1992
Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, Armbrecht AM, Dhillon B, Deary IJ, Redmond E, Bird AC, Moore AT (2007) Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med 357:553–561
Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM (2007) Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet 39:1200–1201
Spencer KL, Olson LM, Anderson BM, Schnetz-Boutaud N, Scott WK, Gallins P, Agarwal A, Postel EA, Pericak-Vance MA, Haines JL (2008) C3 R102G polymorphism increases risk of age-related macular degeneration. Hum Mol Genet 17:1821–1824
Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM (2009) Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet 17:100–104
Ennis S, Gibson J, Cree AJ, Collins A, Lotery AJ (2010) Support for the involvement of complement factor I in age-related macular degeneration. Eur J Hum Genet 18:15–16
Hughes AE, Orr N, Esfandiary H, Az-Torres M, Goodship T, Chakravarthy U (2006) A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet 38:1173–1177
Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, Radeke MJ, Kavanagh D, Richards A, Atkinson J, Meri S, Bergeron J, Zernant J, Merriam J, Gold B, Allikmets R, Dean M (2006) Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med 38:592–604
Scholl HPN, Fleckenstein M, Charbel Issa P, Keilhauer C, Holz FG, Weber BHF (2007) An update on the genetics of age-related macular degeneration. Mol Vis 13:196–205
Haddad S, Chen CA, Santangelo SL, Seddon JM (2006) The genetics of age-related macular degeneration: a review of progress to date. Surv Ophthalmol 51:316–363
Katta S, Kaur I, Chakrabarti S (2009) The molecular genetic basis of age-related macular degeneration: an overview. J Genet 88:425–449
Thakkinstian A, Han P, McEvoy M, Smith W, Hoh J, Magnusson K, Zhang K, Attia J (2006) Systematic review and meta-analysis of the association between complement factor H Y402H polymorphisms and age-related macular degeneration. Hum Mol Genet 15:2784–2790
Scholl HPN, Charbel Issa P, Walier M, Janzer S, Pollok-Kopp B, Borncke F, Fritsche LG, Chong NV, Fimmers R, Wienker T, Holz FG, Weber BHF, Oppermann M (2008) Systemic complement activation in age-related macular degeneration. PLoS One 3:e2593
Reynolds R, Hartnett ME, Atkinson JP, Giclas PC, Rosner B, Seddon JM (2009) Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Invest Ophthalmol Vis Sci 50:5818–5827
Hecker LA, Edwards AO, Ryu E, Tosakulwong N, Baratz KH, Brown WL, Charbel Issa P, Scholl HPN, Pollok-Kopp B, Schmid-Kubista KE, Bailey KR, Oppermann M (2010) Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum Mol Genet 19:209–215
Skerka C, Lauer N, Weinberger AA, Keilhauer CN, Suhnel J, Smith R, Schlotzer-Schrehardt U, Fritsche L, Heinen S, Hartmann A, Weber BH, Zipfel PF (2007) Defective complement control of factor H (Y402H) and FHL-1 in age-related macular degeneration. Mol Immunol 44:3398–3406
Laine M, Jarva H, Seitsonen S, Haapasalo K, Lehtinen MJ, Lindeman N, Anderson DH, Johnson PT, Jarvela I, Jokiranta TS, Hageman GS, Immonen I, Meri S (2007) Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol 178:3831–3836
Ormsby RJ, Ranganathan S, Tong JC, Griggs KM, Dimasi DP, Hewitt AW, Burdon KP, Craig JE, Hoh J, Gordon DL (2008) Functional and structural implications of the complement factor H Y402H polymorphism associated with age-related macular degeneration. Invest Ophthalmol Vis Sci 49:1763–1770
Sjoberg AP, Trouw LA, Clark SJ, Sjolander J, Heinegard D, Sim RB, Day AJ, Blom AM (2007) The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. J Biol Chem 282:10894–10900
Herbert AP, Deakin JA, Schmidt CQ, Blaum BS, Egan C, Ferreira VP, Pangburn MK, Lyon M, Uhrin D, Barlow PN (2007) Structure shows that a glycosaminoglycan and protein recognition site in factor H is perturbed by age-related macular degeneration-linked single nucleotide polymorphism. J Biol Chem 282:18960–18968
Clark SJ, Higman VA, Mulloy B, Perkins SJ, Lea SM, Sim RB, Day AJ (2006) His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J Biol Chem 281:24713–24720
Yu J, Wiita P, Kawaguchi R, Honda J, Jorgensen A, Zhang K, Fischetti VA, Sun H (2007) Biochemical analysis of a common human polymorphism associated with age-related macular degeneration. Biochemistry 46:8451–8461
Tortajada A, Montes T, Martinez-Barricarte R, Morgan BP, Harris CL, de Cordoba SR (2009) The disease-protective complement factor H allotypic variant Ile62 shows increased binding affinity for C3b and enhanced cofactor activity. Hum Mol Genet 18:3452–3461
Zhou J, Jang YP, Kim SR, Sparrow JR (2006) Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci USA 103:16182–16187
Wang AL, Lukas TJ, Yuan M, Du N, Handa JT, Neufeld AH (2009) Changes in retinal pigment epithelium related to cigarette smoke: possible relevance to smoking as a risk factor for age-related macular degeneration. PLoS One 4:e5304
Hollyfield JG, Perez VL, Salomon RG (2010) A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol Neurobiol 41:290–298
Chen H, Liu B, Lukas TJ, Neufeld AH (2008) The aged retinal pigment epithelium/choroid: a potential substratum for the pathogenesis of age-related macular degeneration. PLoS One 3:e2339
Wasmuth S, Lueck K, Baehler H, Lommatzsch A, Pauleikhoff D (2009) Increased vitronectin production by complement-stimulated human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 50:5304–5309
Thurman JM, Renner B, Kunchithapautham K, Ferreira VP, Pangburn MK, Ablonczy Z, Tomlinson S, Holers VM, Rohrer B (2009) Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J Biol Chem 284:16939–16947
Chen M, Forrester JV, Xu H (2007) Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res 84:635–645
Chen M, Muckersie E, Robertson M, Forrester JV, Xu H (2008) Up-regulation of complement factor B in retinal pigment epithelial cells is accompanied by complement activation in the aged retina. Exp Eye Res 87:543–550
Coffey PJ, Gias C, McDermott CJ, Lundh P, Pickering MC, Sethi C, Bird A, Fitzke FW, Maass A, Chen LL, Holder GE, Luthert PJ, Salt TE, Moss SE, Greenwood J (2007) Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc Natl Acad Sci USA 104:16651–16656
Nitsch D, Douglas I, Smeeth L, Fletcher A (2008) Age-related macular degeneration and complement activation-related diseases: a population-based case-control study. Ophthalmology 115:1904–1910
Zipfel PF (2009) Complement and immune defense: from innate immunity to human diseases. Immunol Lett 126:1–7
McAvoy CE, Silvestri G (2005) Retinal changes associated with type 2 glomerulonephritis. Eye 19:985–989
Duvall-Young J, MacDonald MK, McKechnie NM (1989) Fundus changes in (type II) mesangiocapillary glomerulonephritis simulating drusen: a histopathological report. Br J Ophthalmol 73:297–302
Duvall-Young J, Short CD, Raines MF, Gokal R, Lawler W (1989) Fundus changes in mesangiocapillary glomerulonephritis type II: clinical and fluorescein angiographic findings. Br J Ophthalmol 73:900–906
Boon CJ, van de Kar NC, Klevering BJ, Keunen JE, Cremers FP, Klaver CC, Hoyng CB, Daha MR, den Hollander AI (2009) The spectrum of phenotypes caused by variants in the CFH gene. Mol Immunol 46:1573–1594
Sepp T, Khan JC, Thurlby DA, Shahid H, Clayton DG, Moore AT, Bird AC, Yates JR (2006) Complement factor H variant Y402H is a major risk determinant for geographic atrophy and choroidal neovascularization in smokers and nonsmokers. Invest Ophthalmol Vis Sci 47:536–540
Magnusson KP, Duan S, Sigurdsson H, Petursson H, Yang Z, Zhao Y, Bernstein PS, Ge J, Jonasson F, Stefansson E, Helgadottir G, Zabriskie NA, Jonsson T, Bjornsson A, Thorlacius T, Jonsson PV, Thorleifsson G, Kong A, Stefansson H, Zhang K, Stefansson K, Gulcher JR (2006) CFH Y402H confers similar risk of soft drusen and both forms of advanced AMD. PLoSMed 3:e5
Francis PJ, Schultz DW, Hamon S, Ott J, Weleber RG, Klein ML (2007) Haplotypes in the complement factor H (CFH) gene: associations with drusen and advanced age-related macular degeneration. PLoS One 2:e1197
Seddon JM, George S, Rosner B, Klein ML (2006) CFH gene variant, Y402H, and smoking, body mass index, environmental associations with advanced age-related macular degeneration. Hum Hered 61:157–165
Shuler RK Jr, Schmidt S, Gallins P, Hauser MA, Scott WK, Caldwell J, Agarwal A, Haines JL, Pericak-Vance MA, Postel EA (2008) Peripheral reticular pigmentary change is associated with complement factor H polymorphism (Y402H) in age-related macular degeneration. Ophthalmology 115:520–524
Wegscheider BJ, Weger M, Renner W, Steinbrugger I, Marz W, Mossbock G, Temmel W, El-Shabrawi Y, Schmut O, Jahrbacher R, Haas A (2007) Association of complement factor H Y402H gene polymorphism with different subtypes of exudative age-related macular degeneration. Ophthalmology 114:738–742
Leveziel N, Zerbib J, Richard F, Querques G, Morineau G, Fremeaux-Bacchi V, Coscas G, Soubrane G, Benlian P, Souied EH (2008) Genotype-phenotype correlations for exudative age-related macular degeneration associated with homozygous HTRA1 and CFH genotypes. Invest Ophthalmol Vis Sci 49:3090–3094
Brantley MA Jr, Edelstein SL, King JM, Apte RS, Kymes SM, Shiels A (2007) Clinical phenotypes associated with the complement factor H Y402H variant in age-related macular degeneration. Am J Ophthalmol 144:404–408
Andreoli MT, Morrison MA, Kim BJ, Chen L, Adams SM, Miller JW, DeAngelis MM, Kim IK (2009) Comprehensive analysis of complement factor H and LOC387715/ARMS2/HTRA1 variants with respect to phenotype in advanced age-related macular degeneration. Am J Ophthalmol 148:869–874
Leveziel N, Puche N, Richard F, Somner JE, Zerbib J, Bastuji-Garin S, Cohen SY, Korobelnik JF, Sahel J, Soubrane G, Benlian P, Souied EH (2010) Genotypic influences on severity of exudative age-related macular degeneration. Invest Ophthalmol Vis Sci 51:2620–2625
Grassi MA, Folk JC, Scheetz TE, Taylor CM, Sheffield VC, Stone EM (2007) Complement factor H polymorphism p.Tyr402His and cuticular Drusen. Arch Ophthalmol 125:93–97
Boon CJ, Klevering BJ, Hoyng CB, Zonneveld-Vrieling MN, Nabuurs SB, Blokland E, Cremers FP, den Hollander AI (2008) Basal laminar drusen caused by compound heterozygous variants in the CFH gene. Am J Hum Genet 82:516–523
Ciardella AP, Donsoff IM, Huang SJ, Costa DL, Yannuzzi LA (2004) Polypoidal choroidal vasculopathy. Surv Ophthalmol 49:25–37.
Laude A, Cackett PD, Vithana EN, Yeo IY, Wong D, Koh AH, Wong TY, Aung T (2010) Polypoidal choroidal vasculopathy and neovascular age-related macular degeneration: same or different disease? Prog Retin Eye Res 29:19–29
Kondo N, Honda S, Kuno S, Negi A (2009) Coding variant I62V in the complement factor H gene is strongly associated with polypoidal choroidal vasculopathy. Ophthalmology 116:304–310
Lee KY, Vithana EN, Mathur R, Yong VH, Yeo IY, Thalamuthu A, Lee MW, Koh AH, Lim MC, How AC, Wong DW, Aung T (2008) Association analysis of CFH, C2, BF, and HTRA1 gene polymorphisms in Chinese patients with polypoidal choroidal vasculopathy. Invest Ophthalmol Vis Sci 49:2613–2619
Hayashi H, Yamashiro K, Gotoh N, Nakanishi H, Nakata I, Tsujikawa A, Otani A, Saito M, Iida T, Matsuo K, Tajima K, Yamada R, Yoshimura N (2010) CFH and ARMS2 Variations in age-related macular degeneration, polypoidal choroidal vasculopathy, and retinal angiomatous proliferation. Invest Ophthalmol Vis Sci 51:5914–5919
Lima LH, Schubert C, Ferrara DC, Merriam JE, Imamura Y, Freund KB, Spaide RF, Yannuzzi LA, Allikmets R (2010) Three major loci involved in age-related macular degeneration are also associated with polypoidal choroidal vasculopathy. Ophthalmology 117:1567–1570
Farwick A, Wellmann J, Stoll M, Pauleikhoff D, Hense HW (2010) Susceptibility genes and progression in age-related maculopathy: a study of single eyes. Invest Ophthalmol Vis Sci 51:731–736
Despriet DD, Klaver CC, Witteman JC, Bergen AA, Kardys I, de Maat MP, Boekhoorn SS, Vingerling JR, Hofman A, Oostra BA, Uitterlinden AG, Stijnen T, van Duijn CM, de Jong PT (2006) Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. JAMA 296:301–309
Seddon JM, Francis PJ, George S, Schultz DW, Rosner B, Klein ML (2007) Association of CFH Y402H and LOC387715 A69S with progression of age-related macular degeneration. JAMA 297:1793–1800
Baird PN, Robman LD, Richardson AJ, Dimitrov PN, Tikellis G, McCarty CA, Guymer RH (2008) Gene-environment interaction in progression of AMD: the CFH gene, smoking and exposure to chronic infection. Hum Mol Genet 17:1299–1305
Seddon JM, Reynolds R, Maller J, Fagerness JA, Daly MJ, Rosner B (2009) Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Invest Ophthalmol Vis Sci 50:2044–2053
Francis PJ, Hamon SC, Ott J, Weleber RG, Klein ML (2009) Polymorphisms in C2, CFB and C3 are associated with progression to advanced age related macular degeneration associated with visual loss. J Med Genet 46:300–307
Scholl HPN, Fleckenstein M, Fritsche LG, Schmitz-Valckenberg S, Gobel A, Adrion C, Herold C, Keilhauer CN, Mackensen F, Mossner A, Pauleikhoff D, Weinberger AW, Mansmann U, Holz FG, Becker T, Weber BHF (2009) CFH, C3 and ARMS2 are significant risk loci for susceptibility but not for disease progression of geographic atrophy due to AMD. PLoS One 4:e7418
Klein ML, Ferris FL 3rd, Francis PJ, Lindblad AS, Chew EY, Hamon SC, Ott J (2010) Progression of geographic atrophy and genotype in age-related macular degeneration. Ophthalmology 117:1554–1559
Age-Related Eye Disease Study Research Group (2001) A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol 119: 1417-1436
Cho E, Hung S, Willett WC, Spiegelman D, Rimm EB, Seddon JM, Colditz GA, Hankinson SE (2001) Prospective study of dietary fat and the risk of age-related macular degeneration. Am J Clin Nutr 73:209–218
Chong EWT, Kreis AJ, Wong TY, Simpson JA, Guymer RH (2008) Dietary omega-3 fatty acid and fish intake in the primary prevention of age-related macular degeneration — a systematic review and meta-analysis. Arch Ophthalmol 126:826–833
Chua B, Flood V, Rochtchina E, Wang JJ, Smith W, Mitchell P (2006) Dietary fatty acids and the 5-year incidence of age-related maculopathy. Arch Ophthalmol 124:981–986
Klein ML, Francis PJ, Rosner B, Reynolds R, Hamon SC, Schultz DW, Ott J, Seddon JM (2008) CFH and LOC387715/ARMS2 genotypes and treatment with antioxidants and zinc for age-related macular degeneration. Ophthalmology 115:1019–1025
Ho L, van Leeuwen R, Witteman JCM, van Duijn CM, Uitterlinden AG, Hofman A, de Jong PTVM, Vingerling JR, Klaver CC (2010) Can dietary antioxidants reduce the genetic risk of age-related macular degeneration? The Rotterdam Study. Arch Ophthalmol (in press)
Conley YP, Jakobsdottir J, Mah T, Weeks DE, Klein R, Kuller L, Ferrell RE, Gorin MB (2006) CFH, ELOVL4, PLEKHA1, and LOC387715 genes and susceptibility to age-related maculopathy: AREDS and CHS cohorts and meta-analyses. Hum Mol Genet 15:3206–3218
Wang JJ, Rochtchina E, Smith W, Klein R, Klein BE, Joshi T, Sivakumaran TA, Iyengar S, Mitchell P (2009) Combined effects of complement factor H genotypes, fish consumption, and inflammatory markers on long-term risk for age-related macular degeneration in a cohort. Am J Epidemiol 169:633–641
Schaumberg DA, Hankinson SE, Guo Q, Rimm E, Hunter DJ (2007) A prospective study of 2 major age-related macular degeneration susceptibility alleles and interactions with modifiable risk factors. Arch Ophthalmol 125:55–62
DeAngelis MM, Ji F, Kim IK, Adams S, Capone A Jr, Ott J, Miller JW, Dryja TP (2007) Cigarette smoking, CFH, APOE, ELOVL4, and risk of neovascular age-related macular degeneration. Arch Ophthalmol 125:49–54
Hughes AE, Orr N, Patterson C, Esfandiary H, Hogg R, McConnell V, Silvestri G, Chakravarthy U (2007) Neovascular age-related macular degeneration risk based on CFH, LOC387715/HTRA1, and smoking. PLoS Med 4:e355
Schlingemann RO, Witmer AN (2009) Treatment of retinal diseases with VEGF antagonists. Prog Brain Res 175:253–267
Brantley MA Jr, Fang AM, King JM, Tewari A, Kymes SM, Shiels A (2007) Association of complement factor H and LOC387715 genotypes with response of exudative age-related macular degeneration to intravitreal bevacizumab. Ophthalmology 114:2168–2173
Lee AY, Raya AK, Kymes SM, Shiels A, Brantley MA Jr (2009) Pharmacogenetics of complement factor H (Y402H) and treatment of exudative age-related macular degeneration with ranibizumab. Br J Ophthalmol 93:610–613
Goverdhan SV, Hannan S, Newsom RB, Luff AJ, Griffiths H, Lotery AJ (2008) An analysis of the CFH Y402H genotype in AMD patients and controls from the UK, and response to PDT treatment. Eye 22:849–854
Brantley MA Jr, Edelstein SL, King JM, Plotzke MR, Apte RS, Kymes SM, Shiels A (2009) Association of complement factor H and LOC387715 genotypes with response of exudative age-related macular degeneration to photodynamic therapy. Eye 23:626–631
Feng X, Xiao J, Longville B, Tan AX, Wu XN, Cooper MN, McAllister IL, Isaacs T, Palmer LJ, Constable IJ (2009) Complement factor H Y402H and C-reactive protein polymorphism and photodynamic therapy response in age-related macular degeneration. Ophthalmology 116:1908–1912
Tsuchihashi T, Mori K, Horie-Inoue K, Gehlbach PL, Kabasawa S, Takita H, Ueyama K, Okazaki Y, Inoue S, Awata T, Katayama S, Yoneya S (2010) Complement factor H and high-temperature requirement A-1 genotypes and treatment response of age-related macular degeneration. Ophthalmology [Epub ahead of print], doi:10.1016/j.ophtha.2010.04.007
Seitsonen SP, Jarvela IE, Meri S, Tommila PV, Ranta PH, Immonen IJ (2007) The effect of complement factor H Y402H polymorphism on the outcome of photodynamic therapy in age-related macular degeneration. Eur J Ophthalmol 17:943–949
Hakobyan S, Harris CL, Tortajada A, de Goicochea JE, Garcia-Layana A, Fernandez-Robredo P, de Rodriguez CS, Morgan BP (2008) Measurement of factor H variants in plasma using variant-specific monoclonal antibodies: application to assessing risk of age-related macular degeneration. Invest Ophthalmol Vis Sci 49:1983–1990
Kelly U, Rickman CB, Postel EA, Hauser MA, Hageman GS, Arshavsky VY, Skiba NP (2009) Rapid and sensitive method for detection of Y402, H402, I62, and V62 variants of complement factor H in human plasma samples using mass spectrometry. Invest Ophthalmol Vis Sci 50:1540–1545
Mollnes TE, Kirschfink M (2006) Strategies of therapeutic complement inhibition. Mol Immunol 43:107–121
Wagner E, Frank MM (2010) Therapeutic potential of complement modulation. Nat Rev Drug Discov 9:43–56
Charbel Issa P, Tröger E, Finger R, Holz FG, Wilke R, Scholl HPN (2010) Structure–function correlation of the human central retina. PLoS One 5:e12864
Acknowledgements
This work was supported by the European Commission, Seventh European Community Framework Program, Marie Curie Intra-European Fellowship (237238) to PCI; the Wynn-Gund Translational Research Acceleration Program Enhanced Research and Clinical Training Award, National Neurovision Research Institute (NNRI) — Foundation Fighting Blindness (FFB; NNCD-CL-0310.0049-JHU-WG) to HPNS; and the Macular Degeneration Research Award, American Health Assistance Foundation (AHAF; M2010042) to HPNS.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Charbel Issa, P., Victor Chong, N. & Scholl, H.P.N. The significance of the complement system for the pathogenesis of age-related macular degeneration — current evidence and translation into clinical application. Graefes Arch Clin Exp Ophthalmol 249, 163–174 (2011). https://doi.org/10.1007/s00417-010-1568-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-010-1568-6