
Abstract
Sporadic Creutzfeldt-Jakob disease (sCJD) is the commonest human prion disease, occurring most likely as the consequence of spontaneous formation of abnormal prion protein in the central nervous system (CNS). Variant Creutzfeldt–Jakob disease (vCJD) is an acquired prion disease that was first identified in 1996. In marked contrast to vCJD, previous investigations in sCJD revealed either inconsistent levels or an absence of PrPSc in peripheral tissues. These findings contributed to the consensus that risks of transmitting sCJD as a consequence of non-CNS invasive clinical procedures were low. In this study, we systematically measured prion infectivity levels in CNS and peripheral tissues collected from vCJD and sCJD patients. Unexpectedly, prion infectivity was detected in a wide variety of peripheral tissues in sCJD cases. Although the sCJD infectivity levels varied unpredictably in the tissues sampled and between patients, these findings could impact on our perception of the possible transmission risks associated with sCJD.
Similar content being viewed by others


Introduction
Transmissible spongiform encephalopathies (TSEs), or prion diseases, are fatal neurodegenerative disorders that affect a large spectrum of mammalian species. The fundamental event in prion propagation is the conversion of the normal cellular prion protein (PrPC) into an abnormal disease-associated isoform (PrPSc) in the brain and other tissues of infected individuals. PrPC is completely degraded by digestion with proteinase K (PK), whereas PrPSc is N-terminally truncated, resulting in a PK-resistant core termed PrPres [41]. According to the prion concept, PrPSc is the principal, if not the sole, component of the transmissible prion agent [49] and PrPres is a tissue marker for prion diseases [41, 51]. Particular biochemical properties of PrPSc, such as detergent solubility, PK resistance and electromobility evidenced by western blot can be used to distinguish between different prion agents or strains [5, 6]
In humans, Creutzfeldt-Jakob disease (CJD) is a low incidence disease (≈ 1 case per million population per year) that occurs most often as either a sporadic (sCJD) or a familial/genetic (fCJD) form. Whereas familial disease forms are linked to pathogenic mutations in the human prion protein gene (PRNP), no clear epidemiologic risk factors have been identified for sporadic disease forms [57]. In the absence of identifiable external sources that might explain sCJD occurrence, it is currently assumed that this disease is triggered by the spontaneous and stochastic formation of a misfolded PrP nucleus in the brain of the affected individual. This original nucleus is considered to recruit and convert fresh PrPC into PrPSc leading to the progressive spread and propagation of prions in the brain of the diseased patient. In fCJD cases, PRNP mutations foster the formation of the initial misfolded PrP nucleus, leading to an increased probability of development of the disease [14].
In sCJD, two major brain PrPres isoforms have been described by western blot (WB): in type 1 PrPres, the apparent molecular weight of the unglycosylated fragment is 21 kDa, while in type 2, it is 19 kDa [45]. Sporadic CJD is not a uniform disorder in terms of its clinical and neuropathological phenotypes and is subclassified according to the polymorphism at codon 129 of the PRNP sequence (Methionine/Valine) and to the electromobility of the PK resistant core of the abnormal PrP (PrPres: either as type 1 or 2) [45]. The experimental transmission of sCJD tissue isolates in transgenic mice that express human PrP (TgHu) has confirmed that sCJD is associated with a several prion strains that vary in their biological and biochemical properties [7, 11]. Although the complete spectrum of sCJD-associated prion strain remains unknown, there is now significant evidence that the two most frequent categories of sCJD, MM/MV type 1 and MV/VV type 2, are predominantly caused by two distinct prion strains, named M1CJD and V2CJD respectively [7, 11, 12, 25, 33].
In 1996, a new form of CJD, named variant CJD (vCJD), was identified in humans. Variant CJD was demonstrated to be caused by the prion strain responsible for bovine spongiform encephalopathy in cattle, which is distinct from any of the identified sCJD prion strains [29, 30]. Following the emergence of vCJD, it was established that in both affected and asymptomatic patients the vCJD agent accumulated in a large variety of peripheral tissues, including lymphoid organs and blood [15]. On the basis of these findings, the risk of vCJD transmission by surgical and medical procedures has been considered as serious by international health authorities, leading to the implementation of systematic preventive measures aiming at limiting the risk of iatrogenic vCJD transmission [56].
In marked contrast with vCJD, previous investigations carried out in sCJD patients revealed either low or inconsistent levels or an absence of detectable PrPSc in peripheral tissues [24, 28]. These findings led to the general consensus that the risk of transmitting sCJD in the framework of a non-Central Nervous System (CNS) invasive clinical procedure was low.
Recently, bioassays in transgenic mice that express the human PrP gene and display a high sensitivity to sCJD demonstrated the presence of prion infectivity in the plasma and bone marrow of several sCJD patients [17, 32]. These results raised questions about the overall distribution of prion infectivity and transmission risks associated with other peripheral tissues from sCJD patients.
In this study, we measured the prion infectivity levels in a panel of tissues collected from vCJD and sCJD MM1 cases. These bioassays demonstrated that, as expected, consistent titres of infectivity were present in lymphoid tissues from vCJD patients. However, for the non-lymphoid peripheral tissues studied, variable and lower titres of infectivity were detected in both sCJD and vCJD patients. These findings could impact on our perception of the possible transmission risks associated with sCJD involving non-CNS invasive procedures.
Methods
Ethics statement
All animal experiments were performed in compliance with institutional and French national guidelines and in accordance with the European Community Council Directive 86/609/EEC. The animal experiments that are part of this study (national registration 01734.01) were approved by the local ENVT ethic committee. Mouse inoculations were performed under anaesthesia (isofulorane). Mice that displayed clinical signs were anesthetized with isoflurane before sacrifice using CO2 inhalation.
Human tissue samples were obtained from the National CJD Research & Surveillance Unit Brain and Tissue Bank in Edinburgh, UK, which is part of the Edinburgh Brain and Tissue Bank. For the purposes of this study, samples were pseudo-anonymized using a Brain Bank reference number. All cases had informed consent for research and their supply and use in this study in keeping with the East of Scotland Research Ethics Service approval for the Edinburgh Brain and Tissue Bank (16/ES/0084).
CJD and control patients
Tissues from four clinical vCJD cases (referred to in this study as vCJD-1 to vCJD-4) and one asymptomatic vCJD-infected individual who received a transfusion of packed red blood cells from a donor who subsequently died from vCJD [47] (vCJD-AS) were investigated. The vCJD cases were homozygous for methionine at codon 129 (Met/Met129), while the asymptomatic case was heterozygous Methionine/Valine at this particular codon. Similarly, tissues from five sCJD cases were investigated. Cases were selected on the basis of their genotype at codon 129 (Met/Met129), their PrPres Western blot profile (type 1) and availability of peripheral tissues samples. Tissues from one non-CJD case (Met/Met129) were used as control (NC-1). The autopsies were performed in various hospitals across the UK with appropriate facilities for cases of suspected CJD. For this group, the median post mortem interval was 48 h (range 30–72 h).
Basic demographic and medical history details for the vCJD, sCJD and control cases are presented in Table 1. In all cases, informed consent was given to examine the entire PRNP coding sequence in order to exclude pathogenic mutations in the PRNP gene [2, 42].
Tissues samples collection and tissue homogenate preparation
Tables 2 and 3 shows the tissue tested in each patient. Tissues samples were collected post mortem using disposable equipment, snap frozen, and stored at − 80 °C. Strict precautions were undertaken to prevent potential cross contamination of samples during collection, handling and storage. Frozen samples were homogenized in 5% glucose in distilled water in grinding tubes (Bio-Rad) adjusted to 10% (w/v) using a TeSeE™ Precess 48™ homogenizer (Bio-Rad). Homogenates were then filtered through a 20 g needle with a syringe.
Mouse bioassays
vCJD and sCJD tissues bioassay were carried out using mice expressing bovine PrP (tgBov-tg110) and human PrP methionine at codon 129 (tg Met-tg340), respectively. TgMet and tgBov mice expressed approximately fourfold and eightfold more PrP compared to that seen in normal human and cattle brain tissue [12, 13, 44]. These two models were already used to measure prion infectivity in vCJD and sCJD MM1 tissues in previous studies [12, 15, 32].
Tissues from the non-CJD control case were inoculated in both tgBov and tg340 mice.
Groups of 6- to 10-week-old female mice (n = 6) were anesthetized and inoculated with 20 µL of a 10% tissue homogenate in the right parietal lobe using a 25-gauge disposable hypodermic needle. Mice were observed daily and their neurological status was assessed weekly. When clinically progressive TSE disease was evident, the animals were euthanized and their brains harvested. Half of the brain from those animals that had displayed TSE clinical signs was fixed by immersion in 10% formol saline and the other half was frozen at – 20 °C. Tissues from animals found dead were frozen (no formalin fixation was performed). In animals where no clinical signs were observed, mice were killed at the end of their natural life-span (600–750 days). In those cases, survival times reported in the table as > 650 dpi or > 750 dpi.
Infectious prion titre estimates
The infectious titre in a reference 10% weight/vol frontal cortex homogenate from a clinical vCJD case and sCJD cases were established by endpoint titration (intracerebral route) in tgBov and tgMet mice respectively. The infectious titre (LD50/g IC in corresponding mice lines) were estimated by the Spearman–Kärber method [40].
The titre of prion infectivity in vCJD and sCJD case samples were estimated using the method developed by Arnold et al. [4].
Abnormal PrP Western blot (WB) detection
PK resistant abnormal PrP extraction (PrPres) and Western blot were performed on tissues as previously described [35]. Immunodetection was performed using one Sha31 (1 µg/ml) monoclonal PrP-specific antibody, which recognizes the amino acid sequences YEDRYYRE (145–152) [18].
Vacuolar lesion profiles
Haematoxylin–Eosin stained paraffin embedded brain tissue sections were used to establish standardised vacuolar lesion profiles in mice as previously described [20, 21]. Each lesion profile was based on data obtained from a minimum of 3 animals.
Results
vCJD transmission
Four clinical vCJD cases (Met129 homozygous) and one asymptomatic vCJD case (Met129/Val129 heterozygous) were selected on the basis of their clinico-pathological features and PrPres Western Blot profile in the brain (Table 1).
A panel of frozen tissues that included CNS (frontal cortex), and 14 different peripheral tissues (such as primary and secondary lymphoid tissues, endocrine and exocrine glands, gonads, kidney, lung, liver, heart and skeletal muscles) from each of these 5 cases was constituted (Table 2). Each sample (10% tissue homogenates) was inoculated by the intracerebral route (IC) to bovine PrP expressing mice (tgBov n = 6 per sample, 20 µL per mouse); a bioassay model identified in previous studies as a sensitive and robust approach for the detection and the quantification of vCJD infectivity [15, 17].
In all four vCJD affected patients, the inoculation of frontal cortex homogenate resulted in a 100% attack rate disease transmission (Table 2). Only 5 out of the 54 peripheral tissues samples failed to transmit disease in tgBov. Each of the 14 different categories of peripheral tissues caused, at variable extent, occurrence of clinical TSE in tgBov. Based on these transmission results (positive versus absence of transmission), the pattern of vCJD infectivity in peripheral tissues was relatively similar across the four vCJD patients (Table 2).
No TSE clinical signs or PrPres accumulation in the brain was observed in tgBov inoculated with frontal cortex from the asymptomatic vCJD case (> 650 days post inoculation). 8 out of the 13 inoculated categories of peripheral tissues transmitted a disease (lymphoid organs, lung, heart, pancreas and thyroid) in tgBov (Table 2), which indicated a more restricted distribution of the prion infectivity in the organs of this asymptomatic Met/Val129 patient than in the clinically affected Met/Met129 vCJD patients.
The PrPres Western blot profile and the vacuolar lesions profiles observed in mice inoculated with peripheral tissues from both clinical vCJD and asymptomatic patients were identical to those observed in tgBov mice inoculated with the brain of the vCJD affected patients (Fig. 1).
No transmission was observed in tgBov mice that received frontal cortex and peripheral tissues homogenates from a non CJD control patient (Met129 homozygous) (Table 2, Fig. 1).

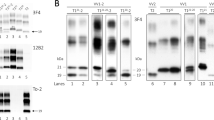
Abnormal PrP and vacuolar lesion profiles in the brain of tgBov mice. Transgenic mice expressing bovine PrP (tgBov) were inoculated with tissues (10% weight/volume homogenates) from 4 vCJD cases (vCJD 1 to 4, homozygous Met129), one vCJD asymptomatic case (vCJD-AS, Met/Val 129 heterozygous) and one non CJD control patient (homozygous Met129) (Table 1). Mice were euthanized when they showed clinical signs of infection or at the end of their natural life-span (600–700 dpi) and their brains were systematically collected. Western-blot and PrPres immunodetection in the brain of tgBov mice inoculated with tissue homogenates a from case vCJD-1 and b the non CJD control case. PrPres Western blots were performed using the Sha31 monoclonal antibody (epitope: 145YEDRYYRE152 of the human-PrP). On each gel (i) a scrapie in sheep isolate (WB cont.) and a vCJD reference isolate were used as controls. Vacuolar lesion profiles in the mice of tgBov mice inoculated with, c frontal cortex from vCJD-1 (empty circle), vCJD-2 (empty inverted triangle), vCJD-3 (empty triangle), vCJD-4 (empty square) and d spleen from vCJD-1 (empty circle), vCJD-2 (empty inverted triangle), vCJD-3 (empty diamond), vCJD-4 (empty square) and vCJD-AS (filled triangle). e Lesion profiles were established in tgBov mice that were scored PrPres positive by western blot following the inoculation with the vCJD-AS case cervical lymph node (empty circle), lung (empty triangle), and spleen (empty square). Lesions profiles were established (using at least 5 individual mice) following the standard method described by Fraser and Dickinson in nine grey matter brain areas (G1 to G9) and three white matter brain areas (W1 to W3) [21]
sCJD transmission
Five sCJD patients were selected on the basis of their clinico-pathological features, genotype at codon 129 of the PRNP gene (Met129 homozygous) and PrPres Western Blot type 1 profile in the brain (MM1 sCJD cases) (Table 1). This is the commonest subtype of sCJD. A panel of 15 peripheral tissues collected from these MM1 sCJD patients was constituted. This panel matched the one investigated in vCJD affected patients (Table 3).
Each sample (10% tissue homogenates) was inoculated by the intracerebral route (IC) to Met129 human PrP expressing mice (tgMet, n = 6 per sample, 20µL per mouse), a mouse model that we already used to detect and quantify prion infectivity in MM1 sCJD patients [32].
The inoculation of 10% frontal cortex homogenates from the sCJD MM1 patients in tgMet (IC route, 6 mice, 20µL per mouse) resulted in a clinical TSE with mean survival times comprised between 200 and 240 days (Table 3). The PrPres WB profile and the vacuolar lesion profile in the brain of the tgMet indicated that a same prion strain was present in the frontal cortex of these five sCJD patients (Fig. 2).

Abnormal PrP and vacuolar lesion profiles in the brain of tgMet mice. Transgenic mice expressing the methionine 129 (TgMet) variant of the human-PRNP were inoculated with tissues (10% weight/volume homogenates) from 5 MM1 sCJD cases and one homozygous Met129 non CJD control case (Table 1). Mice were euthanized when they showed clinical signs of infection or at the end of their natural life-span (> 750 dpi) and their brains were systematically collected. Western-blot and PrPres immunodetection in the brain of tgMet mice inoculated with tissues homogenates a from case sCJD-1 and b the non CJD control case. PrPres Western blots were performed using the Sha31 monoclonal antibody (epitope: 145YEDRYYRE152 of the human-PrP). On each gel (i) a scrapie in sheep isolate (WB cont.) and a MM1 sCJD reference isolate were used as controls. Vacuolar lesion profiles in the mice of tgMet mice inoculated with, c frontal cortex and bone marrow d from sCJD-1 (empty circle), sCJD-2 (empty inverted triangle), sCJD-3 (empty triangle), sCJD-4 (empty square). e Similarly, lesion profiles were established in tgMet mice that were scored PrPres positive by western blot following the inoculation with the sCJD-1 case frontal cortex (filled inverted triangle), lung (empty circle), heart (empty triangle), and salivary gland (empty square). Lesions profiles were established (using at least 5 individual mice), following the standard method described by Fraser and Dickinson in nine grey matter brain areas (G1 to G9) and three white matter brain areas (W1 to W3) [21]
Unexpectedly, in the majority of the cases, the inoculation in tgMet of the peripheral tissues from the same MM1 sCJD patients resulted in positive transmission. TgMet inoculation revealed the presence of prion infectivity in all the different categories of peripheral tissues except liver and gonads (Table 3).
However, in contrast with vCJD, the bioassay results (positive versus absence of transmission) indicated that the prion infectivity distribution pattern in peripheral tissues strongly differed between the five affected MM1 sCJD patients. For instance, in sCJD case 2, positive transmissions were observed in tgMet inoculated with 10 out of the 13 tested peripheral tissues including lymphoid tissues, salivary glands, kidney, heart and pancreas. In contrast, in sCJD case 3, positive transmissions were only observed for 4 out of the same 13 tissues (lung, adrenal gland, bone marrow and skeletal muscle), with no disease transmission resulting from inoculation of lymphoid tissues, salivary gland, kidney, heart or pancreas (Table 3).
Interestingly, bioassay of the lymphoid tissues (spleen, cervical lymph node and tonsil) resulted in a disease transmission in only 3 out of the 5 sCJD cases (cases 2, 4 and 5). These results support the contention that in MM1 sCJD patients, the presence / absence of infectivity in the lymphoid organs is apparently not a determinant driver of the accumulation of infectivity in the other categories of peripheral tissues.
Despite the differences in bioassay transmission patterns, the PrPres Western blot profile and the vacuolar lesions profiles overserved in tgMet mice inoculated with MM1 sCJD peripheral tissues and frontal cortex brain homogenates were identical (Fig. 2), indicating that the same prion strain was present in the peripheral tissues and the CNS of the five MM1 sCJD patients.
No transmission or PrPres accumulation was observed in tgMet mice inoculated with peripheral tissues from the non CJD control patient (Table 3, Fig. 2).
Infectivity titres estimates
In order to estimate the infectivity levels in the vCJD and MM1 sCJD patients’ tissues, we applied the method described by Arnold et al. [4]. This approach uses both the probability of survival (attack rate at each dilution) and the individual mouse survival time at each dilution. The relationship between the titre of inoculum and the probability of infection and the length of the survival times were derived from data corresponding to endpoint titration of a vCJD and a MM1 reference isolate in tgBov and tgMet mice, respectively [17, 32]. A normal distribution for the relationship between dose and survival time was assumed and the probability of infection versus dose was assumed to follow a logistic regression curve (supplementary Fig. 1).
Using this approach, the infectious titre in the frontal cortex of the four vCJD patients was estimated to range between 106.11 and 106.74 ID50 IC in tgBov per gram of tissue (Table 2).
The estimated infectivity levels in secondary lymphoid tissues (spleen and cervical lymph node) were 1 to 4 log10 lower than in the frontal cortex of the same patient. Infectivity levels in the other categories of peripheral tissues were 2.5 to 6 log10 lower than in the frontal cortex (Table 2, Fig. 3).

infectivity levels in peripheral tissues and frontal cortex of vCJD and sCJD patients. Cases vCJD-1 (empty circle), vCJD-2 (empty inverted triangle), vCJD-3 (empty triangle) and vCJD-4 (empty square) died with clinical vCJD. These four patients were Methionine homozygotes at codon 129 of the PRNP gene. Patient vCJD-AS (filled circle) died at an asymptomatic stage of the disease. This patient was heterozygous Methionine/Valine at codon 129 of the PRNP gene. Cases sCJD-1 (empty circle), sCJD-2 (empty inverted triangle), sCJD-3 (empty triangle), sCJD-4 (empty square) and sCJD-5 (empty diamond) died with clinical sCJD. All five were Methionine homozygotes at codon 129 of the PPNP gene. 10% weight/volume tissues homogenates (frontal cortex and peripheral tissues) from vCJD and sCJD cases were prepared and inoculated in bovine PrP (tgBov) and Met129 human PrP expressing mice respectively. Mice were euthanized when they showed clinical signs of infection or after 600 dpi. Mice were considered infected when abnormal PrP deposition was detected in brain by western blot using the Sha31 monoclonal antibody: epitope amino acids 145–152 (YEDRYYRE) of the sheep PrP sequence. Infectious prion titres were estimated using the method developed by Arnold et al. [4]. The model uses both the probability of survival (attack rate at each dilution) and the individual mouse survival times at each dilution to estimate the infectious load
Strikingly, in some of the vCJD affected patients, infectivity levels in heart (vCJD-3 and 4), kidney (vCJD-3 and 4), lung(vCJD-3), salivary gland (vCJD-3 and 4) or thyroid (vCJD-4) were only 1 to 2 log10 lower than in the spleen and/or cervical lymph node (Table 2, Fig. 3).
As already stated, the distribution of the prion in the peripheral tissue of the vCJD asymptomatic case was more restricted than in vCJD affected patients (Fig. 3). However, when positive, the peripheral tissues from the asymptomatic patient displayed similar infectivity levels to those observed in the vCJD patients at the clinical stage of the disease (Table 2, Fig. 3).
In the MM1 sCJD patients, the estimated infectivity levels in the frontal cortex varied between 105.8 and 107.8 ID50 IC in tgMet/gram (Table 3). Infectivity levels in peripheral tissues that scored positive in bioassay were 2.8 to 8 log10 lower than in the frontal cortex of the same patient (Table 3, Fig. 3). In all five cases, no obvious relationship seemed to exist between the infectivity level in the CNS (frontal cortex) and either the distribution or the levels of sCJD infectivity in their peripheral tissues (Fig. 3).
In the sCJD cases 2, 4 and 5, the lymphoid tissues displayed maximal level of infectivity that were 3–3.5 log10 lower than those observed in the frontal cortex (Table 2, Fig. 3). Strikingly, the infectivity levels associated with some peripheral tissues such as salivary gland (sCJD2), heart (sCJD2), kidney (sCJD 2) or bone marrow (sCJD 5) could be equivalent or even higher than those measured in the lymphoid organs of the same patient (Table 2, Fig. 3).
Discussion
vCJD associated risks
Following the emergence of vCJD in the UK in 1996, the presence of abnormal prion protein in the lymphoid tissues of affected patients was rapidly identified [31, 55]. This immediately raised major concerns about the risk of its iatrogenic transmission of the disease (via contaminated surgical instruments and blood transfusion) and led, in many countries, to the implementation of specific preventing against this risk.
Despite a relatively limited number of identified clinical cases (n = 231), the most recent epidemiological studies indicated that 1 out 2000 people in the UK could carry the vCJD agent (as judged by the presence of abnormal prion protein detected by immunohistochemistry in lymphoid follicles in the appendix) and that the exposure period to BSE agent in the UK could have largely exceeded the period initially considered to be at risk (i.e. the 1985–1996 period) [22, 23]. Over the 25 years since the emergence of vCJD only five instances that are a likely consequence of iatrogenic vCJD transmission have come to light, all in the UK and all associated with blood and blood-products [36, 37, 46, 47]. No cases of vCJD due to iatrogenic transmission by medical or surgical procedures have been identified, indicating that the preventive measures implemented to mitigate its transmission were effective [26, 39].
The overall picture of the distribution and levels of prion infectivity in the tissues of both vCJD affected and asymptomatic patients that we here report confirm the numerous hypotheses and the mosaic of experimental data that were used to design the infection control measures that were successful in limiting iatrogenic vCJD transmissions. These results also reinforce the fact that vCJD transmission risks have not disappeared since each asymptomatic vCJD-infected individual in a human population will continue to represent a potential source of disease transmission.
Peripheral tissue infectivity in sCJD
Seminal transmission experiments of peripheral tissues from sCJD cases in primate models failed to detect infectivity in a large selection of peripheral tissues, body fluids and excretions (except in one liver sample) [10]. Abnormal PrP immunodetection techniques (Western blot and immunohistochemistry) also failed to reveal the presence of prion in the peripheral tissues of sCJD patients [27, 28].These findings led to the generally accepted view that prion infectivity in the sCJD remains mostly confined to the CNS.
In 2003, improved Western Blot protocols for PrPres immunodetection revealed the presence of prion in the spleen (10 positive out of 28 cases) and/or the skeletal muscle (8 positive out of 32 cases) [24]. More recently, transmission studies of sCJD tissues in human PrP expressing transgenic mice (using plasma and bone marrow samples) and in vitro amplification of prions in a small number of sCJD peripheral tissues (skin, kidney, lung, adrenal gland) provided further evidence to the view that prions can accumulate in the peripheral tissues of sCJD affected patients [17, 32, 43, 52].
The results that we here report provide unequivocal and definitive evidence of the widespread distribution of the prion infectivity in the peripheral tissues in MM1 sCJD patients.
Other types of patients
Since our study was restricted to MM1 sCJD cases (the commonest sCJD subtype), additional investigations will be necessary to formally establish that consistent accumulation of prions in peripheral tissues also occurs in patients with other PRNP genotypes (Met/Val129 and Val/Val129) and/or affected with other sCJD prion strains [11]. The presence of abnormal PrP (in spleen and the skeletal muscle) and infectivity (in bone marrow) already identified in MV2 and VV2 sCJD patients indicates that this phenomenon is unlikely to be limited to MM1 sCJD cases [24, 32].
Variability in sCJD patients
The nature of peripheral tissues that accumulated infectivity in sCJD and vCJD cases were relatively similar. While the patterns of prion distribution and the infectivity levels observed in the peripheral tissues were relatively homogenous in the vCJD cases, particularly in lymphoid tissues, a high degree of variability was observed across the sCJD cases. Brain vacuolar lesion profiling and PrPres WB typing confirmed that the same prion strain was present in the brain and the peripheral tissues of all the sCJD affected patients. This rules out the hypothesis that prion strain(s) difference(s) (between individuals or in tissues from a same individual) could be responsible for the observed variability.
The comparison of prion distribution pattern in sCJD case 2 (20-months clinical phase duration) and sCJD cases 1, 3, 4 and 5 patients (1–4 months clinical phase duration) might suggest that, at first glance, a longer clinical phase duration is likely to be associated with a more widespread distribution of prions in the body tissues. However, even if our study represents an unprecedented effort for characterizing CJD agent distribution patterns in the organs of affected patients, the number of sCJD cases that we investigated is too limited to draw definitive conclusions. The characterisation of a larger cohort of sCJD patients will be necessary to establish the relationship that might exist between the distribution and/or infectivity levels in peripheral tissues and sCJD patient age at clinical onset and the duration of the clinical phase of the illness.
Iatrogenic transmission of CJD
Several hundred cases of iatrogenic CJD transmission (iCJD) have been reported worldwide, the vast majority of which are likely to represent transmissions from sCJD patients [9]. The principal sources of these outbreaks were intramuscular injections with contaminated human pituitary-derived growth hormone (226 cases) and implantation of dura mater grafts (228 cases) derived from human cadavers with undiagnosed sCJD infections. A small number of cases were apparently also caused by neurosurgery using contaminated neurosurgical instruments and EEG electrodes (6 cases), transplantation of corneal grafts (2 cases) and intramuscular injections with human pituitary-derived gonadotrophic hormone (4 cases) [9].
These cases dramatically illustrate the high resistance of CJD prions to standard medical decontamination procedures and their particular abilities to bind to steel surgical instruments [19, 53]. Survival times in the individuals who were exposed to sCJD agent(s) by the peripheral route could be extremely long and variable; for instance, in patients that received intramuscular injection of contaminated human pituitary-derived growth hormone, the onset of clinical signs and symptoms of iatrogenic CJD could be observed between 4 and 42 years after the treatment [9].
In a context where sCJD infectivity is apparently limited to the CNS, the medical and surgical procedures responsible for iatrogenic transmission of the disease remain relatively limited, the overall risks for sCJD iatrogenic transmission are now considered to be remote since most of these established routes of transmission be avoided (e.g. the use of human pituitary-derived hormones and human dura mater grafts) [9].
Our detection of low levels of sCJD infectivity in non-CNS tissues such as lung, heart, muscle or even salivary gland was unexpected. We next reviewed the medical histories for the sCJD patients that we studied (Table 1), which revealed surgical procedures (s-CJD-2 and 4) and/or invasive medical examinations (polypectomy under colonoscopy sCJD-5) only few years before the clinical disease in these patients. Some years ago, the presence of infectivity in the plasma and the detection of abnormal prion protein in the urine of sCJD patients raised concerns about the risks of sCJD transmission by blood transfusion and plasma/urine-derived medical products [17, 38]. The detection, in our study, of infectivity in the bone marrow and the kidney of sCJD patients further reinforces these concerns.
Prion titres as measured by intracerebral inoculation in PrP over-expressing transgenic mice models can provide us with estimates of the relative infectivity levels present in the CNS and peripheral tissues of affected patients. This approach conforms to the current gold standard for quantification of prions. However, in a context where the amount of infectivity that would be necessary to transmit disease to another human remains unknown, the infectivity titre estimates (as established following intracerebral inoculation in a reporter animal model) cannot be used to directly infer transmission risks to patients. Other factors, including the various different potential exposure routes (subcutaneous, intramuscular, etc.) and the decontamination/sterilisation methodologies used on the surgical instruments /materials involved, will also influence the transmission risk analysis [1].
In many industrialized countries, reliable CJD passive surveillance programs have been established for decades. The apparently stable and low prevalence of sCJD cases in these countries bring some reassurance about the low numbers of iatrogenic CJD cases in this century [9], perhaps reflecting the variable and lower prion titres detected in the non-CNS tissues in this study.
Guidelines are in place to mitigate and control the risk of iatrogenic transmission of CJD in a healthcare setting [1, 56]. The procedures for cleaning and decontamination of surgical instruments and/or medical equipment now includes an assessment of their potential contamination by prions. However, prions are notoriously resistant to physico/chemical treatments and decontamination process that would be efficient on these agents remain generally inapplicable to some surgical and medical equipment [54], resulting in recommendations to destroy neurosurgical instruments that have been used on the brain of a patient with definite or probable CJD. Furthermore, the presence of dementia or an evolving neurodegenerative disorder in a patient undergoing medical or surgical procedures triggers the use of specific protocols designed to prevent the risk of potential transmission of CJD from the equipment used (surgical tools, endoscopes, etc.). The use of cells/tissues/organs and body fluids from these groups of patients for therapeutic purposes (blood donations, tissue grafts, etc.) is also restricted [1, 56].
When considering the risk of iatrogenic transmission of prion agents from CJD patients, one fundamental question is: How early before the occurrence of clinical signs and symptoms is prion infectivity likely to be present in peripheral solid tissues, blood and urine?
All the samples that we used to establish infectivity levels in tissues were collected post mortem in sCJD and vCJD patients at the terminal stage of the disease. There is clear evidence for the presence of infectivity in the blood and the peripheral tissues years before the clinical onset of vCJD in asymptomatic infected patients [3, 8, 16, 17]. However, it is uncertain that the infectivity levels and distribution in the post mortem peripheral tissues of sCJD and vCJD patients reflect the situation that could be observed at a preclinical or early clinical stage in the same patients. In the absence of tissue samples collected from asymptomatic sCJD patients we are totally lacking in data, therefore trying to elaborate further on this question would be totally speculative.
In several forms of genetic prion disease, for example those associated with the E200K PRNP mutation, the clinical disease manifestations, PrPres WB signature and tissue PrPres distributions are similar to that in sCJD [34, 48]. Despite the ethical issues it might raise, the longitudinal collection of blood samples and body fluids, for research purposes, in consenting patients belonging to families affected by these genetic forms of prion diseases (with confirmed mutations of the PRNP gene) may represent the only possibility to address this question.
In conclusion, the systematic surveillance of CJD and related epidemiological studies in many countries confirm the decline of cases of iatrogenic CJD due to recognised medical or surgical procedures, such as human dura mater graft surgery or treatment with human pituitary-derived growth hormone. However, they do not exclude the possibility that iatrogenic transmission could at least partly account for some sCJD cases observed in the population, particularly in localized geographic regions with evidence of CJD case clusters [50].
Many uncertainties remain on the early stages of the prion accumulation and infectivity in the peripheral tissues in patients infected with sCJD. However, the results of this study suggest that the iatrogenic transmission risks associated with sCJD peripheral tissues should not be disregarded.
Data and materials availability
All data are presented in the manuscript and the supplementary information. Human biological material used in the study was provided by the National CJD Research & Surveillance Unit Brain and Tissue Bank in Edinburgh, UK, which is part of the Edinburgh Brain and Tissue Bank supported by the Medical Research Council.
References
ACDP (2015) Transmissible spongiform encephalopathy agents: safe working and the prevention of infection
Andreoletti O, Levavasseur E, Uro-Coste E, Tabouret G, Sarradin P, Delisle MB et al (2002) Astrocytes accumulate 4-hydroxynonenal adducts in murine scrapie and human creutzfeldt-jakob disease. Neurobiol Dis 11:386–393
Andreoletti O, Litaise C, Simmons H, Corbiere F, Lugan S, Costes P et al (2012) Highly efficient prion transmission by blood transfusion. PLoS Pathog 8:e1002782. https://doi.org/10.1371/journal.ppat.1002782
Arnold ME, Hawkins SA, Green R, Dexter I, Wells GA (2009) Pathogenesis of experimental bovine spongiform encephalopathy (BSE): estimation of tissue infectivity according to incubation period. Vet Res 40:8
Bessen RA, Marsh RF (1992) Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol 66:2096–2101
Bessen RA, Marsh RF (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68:7859–7868
Bishop MT, Will RG, Manson JC (2010) Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci USA 107:12005–12010. https://doi.org/10.1073/pnas.1004688107
Bougard D, Brandel JP, Belondrade M, Beringue V, Segarra C, Fleury H et al (2016) Detection of prions in the plasma of presymptomatic and symptomatic patients with variant Creutzfeldt-Jakob disease. Sci Transl Med 8:370ra182. https://doi.org/10.1126/scitranslmed.aag1257
Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, Will RG, Ladogana A et al (2012) Iatrogenic Creutzfeldt–Jakob disease, final assessment. Emerg Infect Dis 18:901–907. https://doi.org/10.3201/eid1806.120116
Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A et al (1994) Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 35:513–529. https://doi.org/10.1002/ana.410350504
Cassard H, Huor A, Espinosa JC, Douet JY, Lugan S, Aron N et al (2020) Prions from sporadic Creutzfeldt-Jakob disease patients propagate as strain mixtures. mBio. https://doi.org/10.1128/mBio.00393-20
Cassard H, Torres JM, Lacroux C, Douet JY, Benestad SL, Lantier F et al (2014) Evidence for zoonotic potential of ovine scrapie prions. Nat Commun 5:5821. https://doi.org/10.1038/ncomms6821
Castilla J, Gutierrez Adan A, Brun A, Pintado B, Ramirez MA, Parra B et al (2003) Early detection of PrPres in BSE-infected bovine PrP transgenic mice. Arch Virol 148:677–691
Colby DW, Prusiner SB (2011) Prions. Cold Spring Harb Perspect Biol 3:a006833. https://doi.org/10.1101/cshperspect.a006833
Douet JY, Lacroux C, Aron N, Head MW, Lugan S, Tillier C et al (2017) Distribution and quantitative estimates of variant Creutzfeldt–Jakob disease prions in tissues of clinical and asymptomatic patients. Emerg Infect Dis 23:946–956. https://doi.org/10.3201/eid2306.161734
Douet JY, Lacroux C, Litaise C, Lugan S, Corbiere F, Arnold M et al (2016) Mononucleated blood cell populations display different abilities to transmit prion disease by the transfusion route. J Virol 90:3439–3445. https://doi.org/10.1128/JVI.02783-15
Douet JY, Zafar S, Perret-Liaudet A, Lacroux C, Lugan S, Aron N et al (2014) Detection of infectivity in blood of persons with variant and sporadic Creutzfeldt–Jakob disease. Emerg Infect Dis 20:114–117. https://doi.org/10.3201/eid2001.130353
Feraudet C, Morel N, Simon S, Volland H, Frobert Y, Creminon C et al (2005) Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem 280:11247–11258. https://doi.org/10.1074/jbc.M407006200
Flechsig E, Hegyi I, Enari M, Schwarz P, Collinge J, Weissmann C (2001) Transmission of scrapie by steel-surface-bound prions. Mol Med 7:679–684
Fraser H, Dickinson AG (1973) Scrapie in mice. Agent-strain differences in the distribution and intensity of grey matter vacuolation. J Comp Pathol 83:29–40
Fraser H, Dickinson AG (1968) The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 78:301–311
Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Brown D, Sinka K et al (2020) Prevalence in Britain of abnormal prion protein in human appendices before and after exposure to the cattle BSE epizootic. Acta Neuropathol 139:965–976. https://doi.org/10.1007/s00401-020-02153-7
Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L et al (2013) Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ 347:f5675. https://doi.org/10.1136/bmj.f5675
Glatzel M, Abela E, Maissen M, Aguzzi A (2003) Extraneural pathologic prion protein in sporadic Creutzfeldt–Jakob disease. N Engl J Med 349:1812–1820
Group E (2001) Genetic epidemiology of Creutzfeldt–Jakob disease in Europe. Rev Neurol (Paris) 157:633–637
Hall V, Brookes D, Nacul L, Gill ON, Connor N, Panel CJDI (2014) Managing the risk of iatrogenic transmission of Creutzfeldt–Jakob disease in the UK. J Hosp Infect 88:22–27. https://doi.org/10.1016/j.jhin.2014.06.002
Head MW, Ritchie D, McLoughlin V, Ironside JW (2003) Investigation of PrPres in dental tissues in variant CJD. Br Dent J 195:339–434. https://doi.org/10.1038/sj.bdj.4810536 ((discussion 331))
Head MW, Ritchie D, Smith N, McLoughlin V, Nailon W, Samad S et al (2004) Peripheral tissue involvement in sporadic, iatrogenic, and variant Creutzfeldt-Jakob disease: an immunohistochemical, quantitative, and biochemical study. Am J Pathol 164:143–153. https://doi.org/10.1016/S0002-9440(10)63105-7
Hill AF, Butterworth RJ, Joiner S, Jackson G, Rossor MN, Thomas DJ et al (1999) Investigation of variant Creutzfeldt–Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 353:183–189
Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J et al (1997) The same prion strain causes vCJD and BSE. Nature 389(448–450):526
Hill AF, Zeidler M, Ironside J, Collinge J (1997) Diagnosis of new variant Creutzfeldt–Jakob disease by tonsil biopsy. Lancet 349:99–100
Huor A, Douet JY, Lacroux C, Lugan S, Tillier C, Aron N, Cassard H, Arnold M, Torres JM, Ironside JW et al (2017) Infectivity in bone marrow from sporadic CJD patients. J Pathol 243:273–278. https://doi.org/10.1002/path.4954
Jaumain E, Quadrio I, Herzog L, Reine F, Rezaei H, Andreoletti O, Laude H, Perret-Liaudet A, Haik S, Beringue V (2016) Absence of evidence for a causal link between bovine spongiform encephalopathy strain variant L-BSE and known forms of sporadic Creutzfeldt–Jakob disease in human PrP transgenic mice. J Virol 90:10867–10874. https://doi.org/10.1128/JVI.01383-16
Kovacs GG, Seguin J, Quadrio I, Hoftberger R, Kapas I, Streichenberger N, Biacabe AG, Meyronet D, Sciot R, Vandenberghe R et al (2011) Genetic Creutzfeldt–Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy. Acta Neuropathol 121:39–57. https://doi.org/10.1007/s00401-010-0713-y
Lacroux C, Comoy E, Moudjou M, Perret-Liaudet A, Lugan S, Litaise C, Simmons H, Jas-Duval C, Lantier I, Beringue V et al (2014) Preclinical detection of variant CJD and BSE prions in blood. PLoS Pathog 10:e1004202. https://doi.org/10.1371/journal.ppat.1004202
Lefrere JJ, Hewitt P (2009) From mad cows to sensible blood transfusion: the risk of prion transmission by labile blood components in the United Kingdom and in France. Transfusion 49:797–812
Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG (2004) Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 363:417–421
Luk C, Jones S, Thomas C, Fox NC, Mok TH, Mead S, Collinge J, Jackson GS (2016) Diagnosing sporadic Creutzfeldt–Jakob disease by the detection of abnormal prion protein in patient urine. JAMA Neurol 73:1454–1460. https://doi.org/10.1001/jamaneurol.2016.3733
Lumley JS, Panel CJDI, Engineering, Scientific Advisory C-P, National Blood Transfusion C, Serious Hazards of Transfusion C (2008) The impact of Creutzfeldt-Jakob disease on surgical practice. Ann R Coll Surg Engl 90:91–94. https://doi.org/10.1308/003588408X261726
Markus RA, Frank J, Groshen S, Azen SP (1995) An alternative approach to the optimal design of an LD50 bioassay. Stat Med 14:841–852
McKinley MP, Bolton DC, Prusiner SB (1983) A protease-resistant protein is a structural component of the scrapie prion. Cell 35:57–62
Moreno CR, Moazami-Goudarzi K, Laurent P, Cazeau G, Andreoletti O, Chadi S, Elsen JM, Calavas D (2007) Which PrP haplotypes in a French sheep population are the most susceptible to atypical scrapie? Arch Virol 152:1229–1232
Orru CD, Yuan J, Appleby BS, Li B, Li Y, Winner D, Wang Z, Zhan YA, Rodgers M, Rarick J et al (2017) Prion seeding activity and infectivity in skin samples from patients with sporadic Creutzfeldt–Jakob disease. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aam7785
Padilla D, Beringue V, Espinosa JC, Andreoletti O, Jaumain E, Reine F, Herzog L, Gutierrez-Adan A, Pintado B, Laude H et al (2011) Sheep and goat BSE propagate more efficiently than cattle BSE in human PrP transgenic mice. PLoS Pathog 7:e1001319
Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ et al (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt–Jakob disease. Ann Neurol 39:767–778
Peden A, McCardle L, Head MW, Love S, Ward HJ, Cousens SN, Keeling DM, Millar CM, Hill FG, Ironside JW (2010) Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia 16:296–304
Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW (2004) Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364:527–529
Poleggi A, van der Lee S, Capellari S, Puopolo M, Ladogana A, De Pascali E et al (2018) Age at onset of genetic (E200K) and sporadic Creutzfeldt-Jakob diseases is modulated by the CYP4X1 gene. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2018-318756
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144
Puopolo M, Catelan D, Capellari S, Ladogana A, Sanguedolce A, Fedele A et al (2020) Spatial epidemiology of sporadic Creutzfeldt–Jakob disease in Apulia, Italy. Neuroepidemiology 54:83–90. https://doi.org/10.1159/000503234
Race R, Raines A, Raymond GJ, Caughey B, Chesebro B (2001) Long-term subclinical carrier state precedes scrapie replication and adaptation in a resistant species: analogies to bovine spongiform encephalopathy and variant creutzfeldt-jakob disease in humans. J Virol 75:10106–10112
Takatsuki H, Fuse T, Nakagaki T, Mori T, Mihara B, Takao M et al (2016) Prion-seeding activity is widely distributed in tissues of sporadic Creutzfeldt–Jakob disease patients. EBioMedicine 12:150–155. https://doi.org/10.1016/j.ebiom.2016.08.033
Taylor D (2002) Inactivation of the BSE agent. C R Biol 325:75–76
Thomas JG, Chenoweth CE, Sullivan SE (2013) Iatrogenic Creutzfeldt–Jakob disease via surgical instruments. J Clin Neurosci 20:1207–1212. https://doi.org/10.1016/j.jocn.2013.01.007
Turner ML, Ironside JW (1998) New-variant Creutzfeldt-Jakob disease: the risk of transmission by blood transfusion. Blood Rev 12:255–268
WHO (1999) WHO infection control guidelines for transmissible spongiform encephalopathies. WHO/CDS/CSR/APH/2000.3
Zerr I, Parchi P (2018) Sporadic Creutzfeldt–Jakob disease. Handb Clin Neurol 153:155–174. https://doi.org/10.1016/B978-0-444-63945-5.00009-X
Acknowledgements
We are grateful to Dr Mark Head for prion protein isotyping, Suzanne Lowrie for scientific and technical support and to Jan Mackenzie for data management and analysis (UK National CJD Research & Surveillance Unit).
Funding
This work was funded by the ‘Fond Européen de Développement Régional, Programme Opérationnel de Coopération Territoriale Espagne France Andorre’ REDPRION (EFA148/16); the UK Food Standards Agency Exploring permeability of the species barrier (M03043 and FS231051); the European Union through FP7 222887 “Priority;”. The National CJD Research & Surveillance Unit is funded by the Department of Health and the Scottish Government. The Edinburgh Brain and Tissue Bank is funded by the Medical Research Council.
Author information
Authors and Affiliations
Contributions
JWI, OA: designed research. JYD, AH, HC, SL, NA, MA, OA: performed research. MA, JMT: contributed new reagents/analytic tools. JYD, DV, JMT, JWI, OA: wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
401_2021_2270_MOESM1_ESM.tif
Dose-response relationship for the survival time and probability of infection of transgenic mice expressing the methionine 129 (Tg Met) of the human-PrP or the bovine PrP (TgBov). Data corresponding to endpoint titration of a vCJD (Met129 homozygous) reference isolate in tgBov (A) and of a MM1 sCJD reference isolate in tgMet (B) were used to derive the relationship between the prion infectivity titre of the inoculum and the probability of infection and the length of the survival time to estimate prion infectivity levels in these two mouse models following the methodology described by Arnold et al [4]. These data have already been used in previous publications [15, 32] (TIFF 8484 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Douet, JY., Huor, A., Cassard, H. et al. Wide distribution of prion infectivity in the peripheral tissues of vCJD and sCJD patients. Acta Neuropathol 141, 383–397 (2021). https://doi.org/10.1007/s00401-021-02270-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-021-02270-x