
Abstract
N-Acetylaspartate (NAA) is the second most abundant organic metabolite in the brain, but its physiological significance remains enigmatic. Toxic NAA accumulation appears to be the key factor for neurological decline in Canavan disease—a fatal neurometabolic disorder caused by deficiency in the NAA-degrading enzyme aspartoacylase. To date clinical outcome of gene replacement therapy for this spongiform leukodystrophy has not met expectations. To identify the target tissue and cells for maximum anticipated treatment benefit, we employed comprehensive phenotyping of novel mouse models to assess cell type-specific consequences of NAA depletion or elevation. We show that NAA-deficiency causes neurological deficits affecting unconscious defensive reactions aimed at protecting the body from external threat. This finding suggests, while NAA reduction is pivotal to treat Canavan disease, abrogating NAA synthesis should be avoided. At the other end of the spectrum, while predicting pathological severity in Canavan disease mice, increased brain NAA levels are not neurotoxic per se. In fact, in transgenic mice overexpressing the NAA synthesising enzyme Nat8l in neurons, supra-physiological NAA levels were uncoupled from neurological deficits. In contrast, elimination of aspartoacylase expression exclusively in oligodendrocytes elicited Canavan disease like pathology. Although conditional aspartoacylase deletion in oligodendrocytes abolished expression in the entire CNS, the remaining aspartoacylase in peripheral organs was sufficient to lower NAA levels, delay disease onset and ameliorate histopathology. However, comparable endpoints of the conditional and complete aspartoacylase knockout indicate that optimal Canavan disease gene replacement therapies should restore aspartoacylase expression in oligodendrocytes. On the basis of these findings we executed an ASPA gene replacement therapy targeting oligodendrocytes in Canavan disease mice resulting in reversal of pre-existing CNS pathology and lasting neurological benefits. This finding signifies the first successful post-symptomatic treatment of a white matter disorder using an adeno-associated virus vector tailored towards oligodendroglial-restricted transgene expression.
Similar content being viewed by others


Introduction
Despite the ill-defined physiological function of N-acetylaspartate (NAA), this metabolite is widely used as a biomarker of neuronal integrity in magnetic resonance spectroscopy [56]. In fact, with CNS concentrations in the millimolar range, NAA is among the most abundant organic metabolites in the mammalian brain and displays a remarkably segmented metabolism. NAA is produced by N-acetyltransferase 8-like (NAT8L) from acetyl-CoA and aspartate in neurons, and hydrolysed into aspartate and acetate by aspartoacylase (ASPA) in oligodendrocytes [60].
Clinical evidence suggests that alterations in NAA metabolism are not well tolerated. The loss of NAT8L activity abolishes NAA production and has been reported for the neurodevelopmental disorder Hypoacetylaspartia, presenting with seizures, ataxia and microcephaly [52]. Conversely, lack of NAA catabolism caused by missense mutations in the ASPA gene results in the leukodystrophy Canavan disease (CD) characterized by a build-up of NAA in brain, blood and urine. CD is a fatal neurodegenerative disorder, where patients fail to reach developmental milestones, and presents with macrocephaly, seizures, widespread CNS vacuolization and hypomyelination [38, 49].
Due to its devastating nature and lack of therapeutic options ASPA gene replacement therapy has been attempted as an experimental treatment for CD patients [48]. The experimental clinical treatment using first generation viral vectors was safe but therapeutic outcomes were marginal. Novel gene therapy options matching requirements for treatment of CD are now becoming available and recombinant adeno-associated virus (AAV) vectors with cell type-specific tropism or ability to cross the blood–brain barrier have been employed in ASPA gene replacement studies [67]. Direct pre-emptive ASPA gene delivery to the neonatal mouse brain utilizing an oligodendrotropic AAV vector prevents CD up to three months of age [25]. Moreover, creating an ectopic NAA sink in neurons or astrocytes using cell type-specific AAV-ASPA vectors was reported to be beneficial in a CD mouse model [2, 31]. These studies indicate that neurological improvements can be achieved by expressing ASPA in any of the major CNS cell types, at least in mouse, a species that is comparably short-lived.
Identifying the role of ASPA in select cell types and tissues will provide a better understanding of the CD pathophysiology and inform on the ideal target tissues for gene therapy. In the CNS abundant oligodendroglial Aspa expression is undisputed, yet additional expression in neurons and microglia has been reported in rats [10, 50, 55]. Aspa is abundantly expressed in peripheral organs [54], and a recent report revealed immune impairments in CD mice [3] indicating that a better understanding of the contribution of extra-cerebral ASPA is required to identify the primary target tissue for therapy. Preclinical gene therapy studies, as well as insights from Nat8l/Aspa double-knockout mice, provide evidence that the aetiology of CD is caused by excess NAA resulting in toxicity rather than an undersupply with NAA-derived acetate causing hypomyelination [2, 32, 51, 69]. These data suggest that targeting NAT8L gene expression to reduce NAA production might present additional therapeutic efficacy for CD.
Here, we used comprehensive phenotyping of novel engineered mouse mutants enabling dissection of the consequences associated with elevated NAA or disrupted ASPA function. These studies showed that in the absence of ASPA, NAA levels correlated with severity of the CD-like pathology. While this germ line gene therapy approach prevented spongiform vacuolisation, complete depletion of NAA elicited abnormal neuronal recruitment and severe startle deficits. At the other end of the spectrum, utilizing mice that overexpress Nat8l in neurons, we demonstrate that increased NAA levels are not neurotoxic per se. Moreover, oligodendrocyte-specific Aspa depletion in conditional mutants (Aspa ∆OL) resulted in endpoints matching those observed in whole body Aspa knockout mice. Despite complete absence of ASPA in the CNS, disease onset was delayed and histopathology was less prominent in Aspa ∆OL animals. This finding has direct therapeutic relevance, because it demonstrates that Aspa expression in peripheral organs lowers central NAA levels thereby protracting disease progression, yet without precluding final stage pathology. Our data also underpin that optimal treatment of CD requires therapeutic ASPA expression in oligodendrocytes, potentially combined with attenuated NAA production.
Finally, we translated our findings preclinically by employing intracranial AAV-ASPA gene replacement therapy targeting oligodendrocytes in post-symptomatic CD mice. This treatment yielded near complete regression of CNS pathology and long-term neurological and neurometabolic benefits. Taken together, this study delivers fundamental new insights in the pathomechanisms underlying CD, and represents the first successful post-symptomatic treatment of a myelin disorder based on an AAV platform engineered for oligodendrocyte-specific transgene expression.
Materials and methods
For additional experimental details please see Supplemental Experimental Procedures.
Animals
ASPA-deficient lacZ-knock-in Aspa lacZ/lacZ mice (AKO) where used to generate Aspa flox/flox mice via Flp-mediated recombination and deletion of the ß-geo cassette as described by [63]. Aspa lacZ and Aspa flox alleles were detected using the genotyping protocol described [54]. Crossing with Cnp Cre/+ mice [45] disrupted the Aspa flox/flox allele within cells of the oligodendrocyte lineage (Aspa ∆OL). The KO-3582 mutant mouse strain with a targeted deletion of the Nat8l gene locus Nat8l tm1(KOMP)Vlcg (NKO) was obtained from the Knockout Mouse Project Repository at UC Davis. Aspa lacZ/lacZ; Nat8l −/− double-knockout mutants were obtained by appropriate crosses between Aspa lacZ/+ and Nat8l −/+ mice. To overexpress Nat8l in CNS neurons in transgenic mice (ThyNAT), the flag epitope-tagged murine Nat8l cDNA [7] was cloned into the Thy-1.2 minigene cassette [12]. The resulting pThy-Flag-Nat8l was linearized and microinjected into C57Bl/6 × B6D2F1 zygotes as described [36]. Offspring used in this study were backcrossed into C57Bl/6 for at least three generations.
AAV vector production and stereotaxic delivery
A cDNA encoding human ASPA was subcloned into an AAV2 plasmid between the 1.3 kb mouse myelin basic protein (Mbp) promoter and the woodchuck posttranscriptional regulatory element (WPRE) followed by a bovine growth hormone poly(A). Packaging, purification and titering of AAV vectors (serotype cy5), was performed as described previously [19, 78]. P30 mice were bilaterally injected with 1 μl containing 2 × 109 vg into each, the striatum (+0.7 mm AP, ± 1.2 mm ML, − 2.6 mm), thalamus (− 1.5 mm AP, ± 1.0 mm ML, − 2.9 mm DV) and cerebellum (− 5.0 mm AP, ± 1.2 mm ML, − 3.1 mm).
Magnetic resonance imaging, 1H-MR spectroscopy and body composition
Mice were anaesthetised using 1.5–2% isoflurane delivered in an 80% O2 and 20% air mixture. Respiration rate was monitored and body temperature maintained at 37 °C throughout the experiment. In vivo MR imaging procedure was performed under general anaesthesia on a 9.4 T Bruker BioSpec Avance III 94/20 magnetic resonance microimaging system (Bruker) equipped with a cryogenic very low temperature, closed cycle cooled RF-coil. Images were acquired with an optimized isotropic 3D T2w Rapid Acquisition with Relaxation Enhancement protocol (TurboRARE) using 8 echoes per echo-train as described previously [39] Whole brain and ventricle structures were segmented using thresholding and delineation methods provided by the 3D slicer package [22]. 1H-MR spectroscopy was performed in 6-months-old male mice. Spectra from a 2 × 2 × 3 mm3 voxel in the thalamus were acquired at an echo time of 10 ms using a PRESS single voxel sequence as described in [54]. Body composition measurements were performed in conscious animals in accordance to the manufacturers instruction using the EchoMRI-900TM with A100 mouse antenna insert (Echo Medical Systems).
Metabolite and myelin lipid profiling
Animals were euthanized at six or nine months, relevant brain regions dissected quickly. Snap frozen brain tissue was pulverized, solvent extracted [46] and the lyophilized and reconstituted aqueous phase assessed using 1H Nuclear magnetic resonance (NMR) spectroscopy on a Bruker AVANCE III HD 600 spectrometer fitted with a cryoprobe (TCI) and refrigerated sample changer. 1H spectra were acquired, both with and without decoupling 13C using bilev composite pulse decoupling, across an effective bandwidth of 48,000 Hz during the acquisition time, on a 30 s duty cycle. Total metabolite pool sizes of lactate, glutamate, GABA, aspartate, glutamine, alanine, NAA, creatine, myo-inositol, taurine, succinate, fumarate, acetate and NAD+ were determined using TOPSPIN (v3.1) from the 1H{13C-decoupled} spectra as described previously [61]. Quantification of NAAG levels was performed from the aqueous fraction of the above extraction and myelin lipids from the lipophilic phase using LC–MS/MS as described [35].
Histology, immunohistochemistry, western blot and qRT-PCR
Standard Hematoxilin & Eosin, Cresyl Violet and Luxol Fast Blue histochemical stainings were performed on paraffin sections and digitized using Mirax (Carl Zeiss) or Aperio (Leica) slide scanners as described previously [79]. Immunofluorescent detection was performed on free-floating sections as described [80] using the following antibodies: rabbit anti-ASPA serum (in house), mouse anti-NeuN (Merck), rat anti-MBP (Abcam), mouse anti-GFAP (Cell Signaling), chicken anti-β-Gal (Abcam), mouse anti-Flag (Cell Signaling) and rabbit anti-Neurofilament 200 (Sigma) followed by the appropriate Alexa-488/594 conjugated secondary antibodies (Thermo Fisher). Immunoperoxidase detection was performed on free-floating sections as described in [15] using mouse anti-APC (Merck) and rabbit anti-NeuN (Cell Signaling), the appropriate biotinolated secondary antibody (Dianova) and a Vectastain Elite ABC kit (Vector Labs). Immunoblotting was performed as described previously [78] with the following antibodies: rabbit anti-ASPA serum, mouse anti-GAPDH (Sigma), rat anti-MBP (Abcam), rat anti-PLP aa3 and rat anti-NG2 (gift of J. Trotter) followed by the appropriate HRP-conjugated secondary antibodies (Dianova). qRT-PCR for Aspa, Nat8l, NAAG synthetase-I and II (Rimklb and Rimkla) and hypoxanthine phosphoribosyltransferase (Hprt) was performed as described [26].
Behavioural testing and brainstem recordings
Ten-week-old mutants and age- and sex-matched wildtype littermates were used for this study. The rotarod, dowel and wire suspension tests were performed as described [33, 54]. Startle testing was conducted in a startle chamber (SR-LAB; San Diego Instruments, San Diego, USA) as described previously [65]. Pre-pulse inhibition (PPI) was calculated as the per cent decrease of the ASR magnitude in trials when the startle stimulus was preceded by a prepulse [100 × (mean ASR amplitude on pulse alone trials − mean ASR amplitude on prepulse-pulse trials)/mean ASR amplitude on pulse alone trials]. Auditory function was assessed by determining auditory brainstem response (ABR) in a sound attenuating chamber as described previously [79]. Flash visual evoked potentials (fVEP) were recorded as described [70].
Statistics
Graphs and statistical analyses were done with GraphPad Prism 6 software (La Jolla, CA). Student’s t test, One-way or two-way ANOVA followed by Holm-Sidak post hoc test was used for statistical analysis as appropriate. Values are presented as the mean ± SEM and p < 0.05 was considered as statistically significant. Multivariate analysis was performed using Simca-P + v11 (Umetrics) as described in [83].
Results
Neurochemical consequences associated with altered NAA metabolism
In order to study the effects of NAA toxicity in our Aspa-deficient lacZ-knock-in mouse line (AKO) [54], we ablated both alleles of the NAA producing Nat8l gene in the AKO line to produce double-knockout (DKO) mice. Nat8l deficient mice (NKO) with functional Aspa expression were utilized to investigate the consequences of NAA depletion and to model hypoacetylaspartia. qRT-PCR analysis of whole brain RNA confirmed absence of Nat8l or Aspa mRNA expression in DKO and the respective single gene mutants (Fig. 1a). NAA was increased in AKO (4.8 ± 0.2-fold) and not detected in NKO and DKO mice (Fig. 1b). NKO and DKO mice were born at Mendelian ratios and generally appeared and developed normally with body weight comparable to wildtype (WT) controls. AKO mice were much lighter at one month of age, caught up by two month, but plateaued from that time onward (Fig. 1c). As NAA has been attributed to lipid turnover and energy expenditure [57], we investigated the effects of genetic NAA imbalances on whole body composition (Fig. 1d). While NAA null NKO mice showed normal muscle and adipose tissue mass, we observed a strong reduction in both lean and particularly fat mass in high NAA AKO. This deficit was restored in DKO mice. The survival rate of AKO and DKO mice dropped steeply during the first 6 weeks of life (Fig. 1e). Beyond this time the maximum longevity of DKO was extended compared to AKO, but despite the lack of an obvious pathology survival did not exceed 600 days. We observed normal longevity of NKO mice.

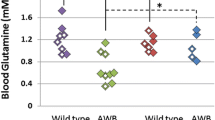
Pathological consequences of manipulation of NAA metabolism. a qRT-PCR analysis of Aspa and Nat8l mRNA expression in CNS tissue from DKO and the parental single gene mutants (n = 3; expression relative to Hprt). b 1H NMR spectroscopy-based quantification of NAA levels in the brainstem of six-month-old WT, AKO, NKO and DKO mice (n = 6). c Body weight development of male WT, AKO, NKO, DKO animals (n = 5–14). d Body composition was determined in nine-month-old male mice using Echo-MRI. Both lean mass (grey) and fat mass (black) was comparable in WT, NKO and DKO mice but fat mass was significantly reduced in AKO (n = 3). e Mean life expectancy of both the AKO (312 days) and DKO (597 days) animals was diminished compared to WT and NKO mice (> 800 days). Note that the number of sudden deaths in AKO and DKO mice was particularly high during the first month of life. f Normalized brain volume obtained from reconstruction of T2-weighted MRIs was increased in AKO but unchanged in NKO and DKO. g Representative 3D reconstruction of T2-weighted MRIs from male WT, AKO, NKO and DKO brains. Segmentation of images was performed in 3D Slicer outlining brain surface (yellow) and ventricles (blue). h 1H NMR spectroscopy-based quantification of metabolite pools in the brain stem from six-month-old WT, AKO, NKO and DKO mice (n = 6). An increase of taurine, inositol, glutamine, creatine and lactate was found in AKO. NKO and DKO showed normal pool sizes except aspartate that was elevated in both lines. i Principal-component analysis of the 1H NMR metabolite profile in WT, AKO, NKO and DKO brainstems was performed on the significantly regulated metabolites excluding NAA. The model accounted for 98% of variance and cross-validated well with a Q2 of 90%. Data represent mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; One-way ANOVA with Holm-Sidak post hoc test
Assessment of high resolution in vivo T2-weighted MRI scans saw no differences in the brain of nine-month-old DKO mice compared to NKO or WT animals. In contrast, AKO mice presented with an increase in both whole brain and ventricle volume (Fig. 1f, g). In line with this finding and concomitant to the observed NAA elevation, we detected a notable increase of organic osmolytes including myo-inositol, taurine and glutamine as well as the important energy metabolites lactate and creatine in the brainstem of AKO mice using 1H-NMR spectroscopy (Fig. 1h). Multivariate analysis including all pools, except NAA revealed a robust segregation of AKO from the other three groups (Fig. 1i). The NKO and DKO cohort clustered differently from WT, which could be attributed largely to a distinct elevation in the aspartate pools in these strains. Substantially decreased numbers of neurons and oligodendrocytes were detected in the thalamus of AKO, a region that shows a high degree of vacuolization (Fig. S1). Cell counts of NKO and DKO compared to controls.
Our initial findings of a near normal phenotype of our DKO mutants suggested that the NAA deletion is beneficial in CD. We then generated AKO mice with only one Nat8l allele and confirmed that the CD-associated pathology can be modulated from severe to mild by varying NAA levels in vivo (Fig. S2).
NAA-deficiency results in neurophysiological abnormalities
First line phenotyping of our DKO mice suggested that disease is prevented if NAA is abolished. However, for a more comprehensive assessment of the translational potential of these results we sought to investigate the neurophysiological consequences of NAA depletion. We employed flash visual evoked potentials (fVEPs), a technique that has been instrumental in identifying dysfunction of the optic system in hypomyelinating conditions [47]. Trace analysis focused on the N3 negative wave (at 52.2 ± 1.4 ms) and the P5 peak (at 98.2 ± 1.4 ms) (Fig. 2a). Indeed, peak latencies were significantly increased in AKO but unchanged from WT in NKO and DKO (Fig. 2b). The restored fVEP latencies in DKO mice confirmed normal myelination of optic nerve and optic tract. These results support our histological and biochemical data on preserved myelination in this line (Fig. S2). Importantly, quantification of the N3 peak amplitude not only revealed a decrease in AKO but also for NKO and DKO, compared with WT controls (Fig. 2c). Reduction of the P5 amplitude in NKO and DKO also approached statistical significance. A decrease in fVEP amplitudes can be attributed to a reduction in the number or sensitivity of neurons in the retinocortical pathway and is an indicator of visual impairments.

Neurophysiological consequences of Nat8l-deficiency. a Representative fVEP traces highlighting the location of two principal peaks N3 and P5 in WT, AKO, NKO and DKO mice. b Quantification of peak latencies assessed in a showed an increase in AKO but no change in NKO and DKO (n = 6). c Quantification of fVEP amplitudes assessed in a showed a decrease in all groups but did not reach statistical significance for NKO and DKO at P5. d Representative traces of the auditory brainstem response (ABR) in 10-week-old mice (n ≥ 6) following a 16 kHz tonepip. Note the absence of waves P4 and P5 in AKO mice, which was restored in DKO. e Quantification of wave peak latencies assessed in d showed an increase for P2 and P3 in AKO. P4 and P5 were completely abolished in AKO. Latencies were unchanged in NKO and DKO. f Input–output growth functions of N2 - P3 from the ABR data shown in d. Linear regression analyses are depicted for stimulus levels between 25 and 60 dB SPL. g Acoustic startle response of 10-week-old mice revealed severe deficits in startle amplitude of AKO (n = 5), NKO (n = 12) and DKO animals (n = 17) compared to WT littermates (n = 28). h Attenuated pre-pulse inhibition (PPI) in NKO and DKO mice compared with controls. Data represent mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; One-way ANOVA with Holm-Sidak post hoc test. SPL sound pressure level, fVEP flash visual evoked potentials
We then investigated sound-evoked auditory brainstem responses (ABR) that are known to have a short latency of 5 ms in controls (Fig. 2d). In AKO mice hypomyelination along the central auditory pathway has been reported to cause substantial delays in mid- to late-ABR waves [79]. Analysis of the ABR waveform stimulated by a 16 kHz tonepip in AKO confirmed increased latency of waveform peak 2 and 3 and absence of peak 4 and 5 compared to WT. NKO and DKO showed no alterations in peak latencies compared with controls, again reflecting normal myelination (Fig. 2e). However, the dynamic range (growth function) of the ABR was significantly decreased in DKO and NKO groups, suggesting that NAT8L-deficiency causes impaired neural recruitment in the auditory brainstem pathway. No significant differences were observed in the AKO group dynamic range compared with WT controls (Fig. 2f).
To further study sensorimotor information processing along the ponto-medullary brainstem axis towards motor neurons, we examined the acoustic startle response (ASR). NKO, AKO and DKO mice alike presented with a severely diminished startle reflex, compared to WT controls (Fig. 2g). While in AKO ASR reduction could be attributed to increased latency in auditory processing (Fig. 2e) and impaired locomotor performance (Fig. S2g), NKO and DKO performed normally in both aspects. In contrast to other facets of the CD pathology that were restored in DKO, the ASR was greatly diminished in DKO and NKO mice. Moreover, the number and appearance of giant neurons of the caudal pontine reticular nucleus, a critical relay station in the ASR pathway, was unchanged (Fig. S3). As anxiety is known to modulate the ASR amplitude [41], we employed the elevated-plus maze test, but did not observe differences between NKO, DKO and WT (not shown). We then assessed sensorimotor gating in the pre-pulse inhibition (PPI) paradigm and observed attenuated PPI amplitudes in NKO and DKO indicating cognitive or psychiatric impairment frequently observed in neurodegenerative disorders [75] (Fig. 2h).
Transgenic mice with increased brain NAA levels are neurologically normal
Our data indicated that neither lack of, nor excess NAA is tolerated. To investigate the cellular compartment most vulnerable to NAA toxicity, we generated transgenic animals with extra copies of the Nat8l gene under the neuronal Thy-1 promoter, in the Aspa WT background. Animals in this line (ThyNAT) were born according to Mendelian ratios and expressed the Flag epitope-tagged NAT8L protein specifically in the brain, but not in other organs (Fig. 3a). Histology revealed Flag-immunoreactivity specifically in the cytosol of CNS neurons (Fig. S4a). Numbers of thalamic neurons and oligodendrocytes were unchanged in ThyNAT compared with controls (Fig. S4b, c). A 1.5-fold Nat8l mRNA increase in brain lysates of ThyNAT mice compared with WT littermates was confirmed by qRT-PCR. Rimkla/b genes mediating NAAG synthesis and Aspa mRNA levels remained unchanged, indicating no substrate feed-forward inhibition at transcriptional level (Fig. 3b). Enhanced Nat8l mRNA expression translated in a 1.78 ± 0.07 and 1.47 ± 0.05 fold increase of NAA concentrations in cortical and brain stem extracts of ThyNAT mice (Fig. 3c). This was paralleled by depleted aspartate pools, reflecting increased incorporation into NAA. Osmolytes that were co-regulated with supra-physiological NAA levels in AKO, such as myo-inositol and taurine (Fig. 1h), were unchanged in ThyNAT mice. Myelination appeared normal and we did not observe vacuolization or astrocytosis (Fig. 3d). We found comparable NAAG concentrations in ThyNAT and controls (Fig. 3e) and unchanged amounts of the major myelin lipid galactosylceramide, further indicating normal myelination (Fig. 3f). Interestingly, we noticed a reduction in size and body weight starting at four weeks and persisting for at least one year (Fig. 3g). This was accompanied by a reduction in both lean and fat mass (Fig. 3h). Motor behaviour (Fig. 3i), startle amplitude (not shown) and sensorimotor gating (Fig. 3j) were normal in ThyNAT mice. The absolute cortical NAA levels in AKO correlated with tissue damage (as a function of neurotoxicity). In contrast, ThyNAT do not show signs of neurotoxicity even though they have higher absolute NAA levels in the cortex than AKO (Fig. S5). These results suggest that elevated NAA levels are uncoupled from neurological dysfunction and pathology.

Neuronal NAT8L overexpression results in elevated NAA levels but does not cause brain damage. a Representative immunoblots confirming that NAT8L-flag transgene expression is restricted to the CNS. In ThyNAT mice NAT8L-flag expression is abundant in cortex (ctx), hippocampus (hc), thalamus (tha), brainstem (bs) and cerebellum (cb). The levels of ASPA, MBP or PLP appear unchanged in ThyNAT whole brain extracts (n = 3). b qRT-PCR confirms the increase of Nat8l mRNA expression in ThyNAT animals compared to WT. Expression of key genes associated with NAA metabolism was not significantly affected. c 1H NMR analysis confirmed increased NAA concentration in ThyNAT cortex and brainstem alongside a reduction in aspartate and creatine levels. Organic osmolytes, including taurine and myo-inositiol, remained unchanged. d Representative H&E/LFB histology (top) shows absence of vacuolization and normal appearance of myelin in ThyNAT mice. GFAP immunohistochemistry (bottom) revealed absence of astrogliosis in transgenic mice. e NAAG concentration in ThyNAT brain stem was comparable to controls. f Cortical concentrations of the major myelin lipid galactosylceramide were similar in transgenic and controls. g Body weight development of male ThyNAT animals. Starting from two months of age, transgenics remained significantly lighter than WT littermates (n = 3–10). h Body composition was determined in nine-month-old, male mice using Echo-MRI. Lean and fat mass was reduced in transgenic animals (n = 6). i Motor performance assessed in three independent tests was unchanged in three-month-old male ThyNAT mice (n ≥ 6). j PPI of the acoustic startle response was normal in 10-week-old, male ThyNAT mice (n = 5) compared with WT (n = 4). Data represent mean ± SEM. **p < 0.01; ***p < 0.001 compared with WT; Student’s t test
Selective Aspa deletion from oligodendrocytes causes Canavan disease
Based on the lack of an overt molecular or cellular phenotype in ThyNAT mice in which NAA accumulation is restricted from oligodendrocytes due to normal ASPA function, we next investigated whether increasing NAA levels in the CNS, by deletion of the Aspa gene exclusively in oligodendrocytes, triggers a CD-like pathology. To address this, we developed mice with a conditional deletion of the Aspa gene exclusively in oligodendrocytes (Aspa ∆OL) by crossing 2′,3′-cyclic nucleotide 3′ phosphodiesterase (Cnp) Cre recombinase knock-in mice (CNP-Cre) with conditional-ready mutant mice in which the second exon of the Aspa gene was flanked by loxP Cre binding sites (Aspa fl/fl). Aspa ∆OL mice were born at the expected Mendelian ratio. Histological examination revealed that ASPA-immunoreactivity was completely abolished in the Aspa ∆OL brain. There was no compensatory ectopic regulation in any other cell type in the CNS (Fig. 4a). Widespread astrogliosis even in the striatum, a brain region not affected by vacuolization, indicated pathology. Immunoblot analysis confirmed that oligodendroglial Aspa deletion in Aspa ∆OL mice abolished ASPA expression throughout the brain while ASPA expression in peripheral organs, including the kidney, remained intact. PLP and MBP protein levels were reduced in Aspa ∆OL mice, suggesting demyelination (Fig. 4b). Quantification of metabolite pools in the brainstem of Aspa ∆OL mice showed a marked increase in NAA, myo-inositol and glutamine (Fig. 4c). Hypomyelination was confirmed by reduced galactosylceramide amounts in brain homogenates (Fig. 4d) and reduced myelin staining in histological sections (Fig. 4e). In the thalamus of Aspa ∆OL mice, a brain region showing pronounced vacuolization, numbers of both neurons and oligodendrocytes were reduced compared with Aspa fl/fl (Fig. S6). In comparison to age-matched AKO mice (Fig. S2d), Aspa ∆OL animals showed relatively moderate vacuolization, even at nine months of age. The physical development of Aspa ∆OL mutants assessed by body weight monitoring was normal up to six months, but was followed by attenuated weight gain compared to age-matched controls (Fig. 4f). Matching our observation in the AKO line (Fig. 1e), longevity of Aspa ∆OL mice was limited to 14 months. However, we did not observe premature fatalities around three weeks of age (Fig. 4g) suggesting a role of peripheral ASPA for juvenile survival. Motor performance, tested between three months and one year, was progressively impaired in Aspa ∆OL compared with controls (Fig. 4h). Compared with AKO, severity of motor deficits in Aspa ∆OL mice was milder up to six months but converged afterwards when Aspa ∆OL mice also presented with paralysis and seizures. NAA levels in the kidney were below the detection limit in WT or Aspa f/f controls and were detectable at low levels in Aspa ∆OL samples (Fig. 4i). In contrast, benchmark AKO samples showed a substantial NAA peak. These findings provide evidence that centrally derived NAA can be degraded by ASPA expressed in peripheral organs. In summary, compared with the whole body knockout, ablating ASPA in oligodendrocytes results in a CD model with delayed progression and partially attenuated late-stage severity.

Selective ASPA depletion in oligodendrocytes causes CD with delayed progression. a Immunohistochemical staining for NeuN (red) and ASPA (green) in the striatum, thalamus and cerebellum of four-month-old Aspa ΔOL animals confirming the complete absence of ASPA throughout the CNS. GFAP immunohistochemistry revealed severe reactive astrogliosis in ASPAΔOL mice but not in controls. b Representative immunoblots showing that ASPA expression was abolished in protein lysates from six-month-old Aspa ΔOL brains (n = 3), but was preserved in the periphery. The central loss of ASPA is associated with reduced expression of the myelin markers PLP-1 and MBP. c Pool sizes of NAA, myo-inositol and glutamine were increased in cortex and brain stem of nine-month-old Aspa ΔOL mice (n = 10) compared with ASPAfl/fl controls (n = 6). d Reduced galactosylceramide (Gal-Cer) concentrations were observed in the cortex of Aspa ΔOL mice (n ≥ 6). e H&E/LFB histology confirmed vacuolization and hypomyelination in 9-months-old Aspa ΔOL mice. f Body weight development in female Aspa ΔOL and Aspa fl/fl mice. Following normal development until 6 months of age the body weight of Aspa ΔOL mice plateaued and remained at reduced levels compared to controls (n = 5–18 per time point). g Reduced longevity of Aspa ΔOL mice. h Motor performance was assessed in the wire suspension test, the dowel test and the rotarod at three, six and nine months. Impaired motor skills of Aspa ΔOL mice (n = 4–14) compared with controls (n ≥ 8) were observed in all tests. Data represent mean ± SEM. **p < 0.01; ***p < 0.001; Student’s t test. i Representative 1H NMR spectra of kidney from Aspa ΔOL, WT and AKO (n = 3). The arrowhead indicates the NAA peak at 2.01 ppm
Preclinical CD gene therapy targeted to symptomatic oligodendrocytes
The data we obtained by phenotyping different CD mouse models showed that oligodendroglial ASPA acts as a natural safeguard against NAA toxicity in the CNS. Next we aimed at translation of our findings for development of a preclinical CD gene therapy based on ASPA gene delivery to oligodendrocytes. As a result of characterizing early disease progression in AKO mice, we identified that supra-physiological levels of NAA and other metabolites, brain vacuolization and hypomyelination manifested at p30 (Fig. S7). To achieve clinical relevance we selected this symptomatic stage as time point of intervention.
We have previously shown that driving oligodendrocyte-specific transgene expression in the CNS is possible using the recombinant mouse myelin basic protein promoter (Mbp) in adeno-associated viral (AAV) vectors [29, 80]. We applied this platform to deliver the human ASPA cDNA (GT mice) or green fluorescent protein (GFP) by direct multi-site injections to AKO followed by longitudinal phenotyping (Fig. 5a). Immunoblotting and qRT-PCR confirmed stable and abundant expression of transgenic ASPA in the CNS of GT mice (Fig. 5b, c). In contrast, virtually no expression of transgenic ASPA was observed in peripheral organs. Immunohistochemical staining corroborated ASPA expression in all target regions and a robust vector spread (Fig. 5d) comparable to our previously published results [80]. Identification of transgene expressing cells in the target areas confirmed approximately 80% oligodendrocyte specificity in the striatum and thalamus alike (Fig. 5e, f; Fig. S8) indicating that at least early during the CD pathology specificity of transgene expression and transduction efficiency does not depend on the tissue integrity at the time point of vector delivery. While the cerebellum was not analysed quantitatively, we approximated equal numbers of hASPA expressing neurons and oligodendrocytes in this region. ASPA was detected in oligodendrocytes of myelinated tracts such as the corpus callosum and fimbria suggesting penetration of the virus vector into compact white matter (not shown).

Long-term oligodendroglial ASPA expression following post-symptomatic AAV delivery. a Schematic overview of the preclinical cy5-Mbp-ASPA gene replacement therapy. b Immunoblot confirming stable recombinant ASPA expression in the CNS of AKO-GT (AAV-Mbp-ASPA) mice compared to AKO-GFP (AAV-Mbp-GFP) control mice at 9 month of age and virtually no expression in the AKO liver following intracranial therapeutic AAV delivery. c qRT-PCR detection of human ASPA mRNA expression in the CNS of nine-month-old cy5-Mbp-ASPA treated AKO mice. Transgenic ASPA mRNA expression was minimal in the liver and kidney. d Immunohistochemical detection of vector spread achieved by intracranial delivery of 2 × 109 vg cy5-Mbp-ASPA to the striatum, thalamus and cerebellum of P30 AKO mice. Percentage of β-Gal+ oligodendrocytes, NeuN+ neurons and GFAP+ astrocytes expressing transgenic human ASPA (hASPA) in the e striatum and the f thalamus of AKO mice eight months following AAV delivery (n = 3). Data represent mean ± SEM. ***p < 0.001; One-way ANOVA with Holm-Sidak post hoc test
Post-symptomatic oligodendroglial ASPA gene therapy reverts Canavan disease
High resolution T2-weighted MRI 5 months following intracranial Mbp-ASPA gene therapy revealed that key aspects of the CD histopathology were reverted in AKO-GT mice (Fig. 6a). Representative in vivo 1H-MR spectra measured in the thalamus of these animals indicated normalisation of the metabolic profile (Fig. 6b). Quantification of brain and ventricle volume revealed that GT normalized both brain volume and excessive ventricle dilation (Fig. 6c). Quantitative analysis of 1H-MR spectra confirmed restoration of excessive NAA and NAAG concentration, normalization of organic metabolite levels exemplified by myo-inositol, taurine and the macromolecular and lipid fraction, as well as replenishment of the glutamate and GABA neurotransmitter pools (Fig. 6d). Neurochemical restoration in GT mice was corroborated by attenuation of brain damage (Fig. 7a). Compared to AKO-GFP controls, in AKO-GT mice numbers of both neurons and oligodendrocytes were restored in the thalamus, a target site for AAV-ASPA delivery (Fig. S9). Immunohistochemical assessment affirmed absence of reactive astrogliosis and microgliosis in AKO-GT mice (Fig. 7b), as well as improved myelination (Fig. 7c). We repeatedly assessed locomotor behaviour employing hanging wire, dowel and rotarod tests and found long-term improvements in GT mice (Fig. 7d).

AAV-Mbp-ASPA-mediated gene therapy in AKO oligodendrocytes restores NAA levels and brain size. a Representative 3D reconstruction of T2-weighted MRIs from male WT, AKO-GFP and AKO-GT mice. Segmentation of images was performed in 3D Slicer outlining brain surface (yellow) and ventricles (blue). b Representative traces of 1H-MR spectroscopy obtained from the above animals. c Normalized brain volume and ventricle size obtained from reconstructed T2-weighted MRIs in AKO-GT animals compared to AKO-GFP controls. d 1H-MR spectroscopy-based quantification of metabolite pools relative to total creatine in the brain stem from six-month-old WT, AKO-GFP, and AKO-GT mice (n = 3). Note, the mm09 refers to a macromolecular and lipid fraction with chemical shift of 0.9 ppm. Data represent mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; One-way ANOVA with Holm-Sidak post hoc test

Post-symptomatic intracranial gene therapy targeting oligodendrocytes improves histopathology and motor performance in CD mice at nine months. a Representative H&E/LFB histology of the cerebellum, hippocampus, thalamus and striatum (from top to bottom) of WT, AKO-GFP, AKO-GT animals. b Representative immunohistochemical staining in the striatum showing normalization of astrogliosis (GFAP; top) and microgliosis (IBA1; bottom) in GT-treated mice. c ASPA gene therapy improves myelination assessed by normalized MBP immunoreactivity in the cerebellum. d Attenuation of weight of aged AKO following intracranial cy5-Mbp-ASPA gene therapy. e Motor performance was assessed at three, six and nine months. Sustained improvement in the wire suspension test, dowel test and on the rotarod approaching WT levels was measured in AKO-GT animals compared to GFP controls (n = 4–30). Data represent mean ± SEM. **p < 0.01; ***p < 0.001; #p < 0.05 (AKO-GFP vs AKO-GT). One-way ANOVA with Holm-Sidak post hoc test
Discussion
Lack of NAA causes neurological dysfunction
Recent studies have indicated that Nat8l could be a therapeutic target for the treatment of CD. This concept prompted a detailed investigation of the consequences associated with altered Nat8l expression in vivo. As expected, Nat8l-deficiency in NKO and DKO mice resulted in the complete loss of NAA and its derivative NAAG. This indicates that NAA, while likely to contribute to osmoregulation, myelination, energy and pH homeostasis in the brain [56, 66, 68, 77], is not essential for these processes. However, when employing tests of sensorimotor function and neuronal recruitment, we identified hitherto unreported deficits in NAA-deficient lines. The notable reduction in the auditory startle response despite normal motor behaviour and functional auditory processing indicates a possible role for NAA in the limbic pathways protecting the body from predatory attacks or sudden trauma. Researching the exact underlying course of action by which NAA-deficiency causes reduced neuronal fibre recruitment following audio-visual stimulation and ASR attenuation was outside the scope of this study. However, our data suggest that aberrant aspartate- or NAAG-dependent neuromodulation may underlie dysfunction at the neurocircuitry level. The increased aspartate pool size resulting from absence of NAT8L can be explained by substrate accumulation. This shift may affect the malate–aspartate shuttle causing alterations in subcellular glutamate localisation and neuronal energy homeostasis. In fact, lack of the neuronal aspartate–glutamate carrier is associated with hyperreflexia [82]. In addition, aspartate can influence the excitatory function of GABAergic cells and promote GABA release via the selective activation of NMDA receptors [43]. Imbalances of GABAergic and glycinergic neurotransmission in axons of giant neurons of the pontine reticular formation, as well as loss of these cells have been associated with impaired ASR [41], and NMDA receptor activity is critical for a normal ASR response [37]. Attenuated PPI, observed in the NKO and DKO lines, has been linked to neuropsychiatric disorders including schizophrenia. These sensorimotor gating deficits may be caused by dysregulation of the dopamine system which has previously been reported for Nat8 l-deficient mice [41, 62, 73].
Elevated NAA concentrations are not intrinsically neurotoxic
Absence of ASPA, causing a detrimental increase of the organic metabolite NAA within oligodendrocytes is the root cause for the neurochemical changes, histopathology and motor deficits observed in CD. Utilizing alternative models, we confirm that key aspects of the pathology are paralleled by dose-dependent NAA toxicity and that NAA depletion prevents CD [32, 51]. Metabolic profiling disclosed a substantial increase in principal osmolytes in Aspa depleted mice that was proportional to the observed spongiform vacuolisation, ventricle size and macrocephaly. In the brain, cell volume has been proposed to be predominantly regulated by organic osmolytes because their flux, in contrast to ions, does not directly perturb the membrane potential and thus electrical activity [5]. Pools of these osmolytes are primarily located in glia and are directly involved in the regulation of water homeostasis, membrane stability, pH and survival [8, 14, 16, 21, 23, 34]. NAA distribution is dynamically regulated during osmotic fluctuations, but the role of NAA as osmolyte remains controversial [6, 8, 18, 30, 64, 72, 74, 76]. Importantly, our findings indicate that increased NAA concentrations are uncoupled from CNS pathology and neurological dysfunction. Direct comparison of cortical NAA elevation to equivalent levels either by neuronal NAT8L overexpression (ThyNAT), or Aspa deletion, provides evidence that NAA toxicity depends on the lack of its oligodendroglial degradation. This suggests that intact ASPA in oligodendrocytes of ThyNAT mice maintains oligodendrocyte NAA at physiological concentrations both intracellularly and in the myelin compartment, thereby preventing disease. Consequently, the increased overall NAA levels in ThyNAT brain extracts reflect increased extracellular or intra-neuronal concentrations that are not toxic per se and do not trigger a substantial alteration in principal osmolytes. This is supported by recent findings that extracellular elevation of NAA does not cause vacuolating histopathology [4]. While the aetiology is multifactorial, based on our data, we hypothesize that the CD pathology is triggered by oligodendroglial intolerance to supra-physiological NAA concentrations. Increased NAA may be particularly detrimental to oligodendrocytes by inducing osmotic challenges to a cell designated to maintain a highly lipid-rich environment or by inducing oxidative stress and lipid peroxidation [24]. Additionally, challenges to maintain energy homeostasis might contribute to the aetiology. NAA-derived aspartate is metabolised by oligodendrocytes where it is thought to contribute to the import of reducing equivalents into mitochondria and enter the tricarboxylic acid cycle [1]. Because NAA is not metabolised, AKO oligodendrocytes might thus experience sustained dysregulation of energy homeostasis. Irrespective of whether oligodendroglial NAA toxicity, lipid peroxidation, or challenges maintaining intracellular osmotic or energy homeostasis are the root cause, the CD pathology is exacerbated by a secondary increase of organic osmolytes involving astrocytes.
While this study confirms that NAA is not essential for normal myelination, the incorporation of NAA-derived acetate into myelin lipids and thus its contribution to myelin synthesis is undisputed [9, 13, 17, 32, 49, 51, 53, 81]. The complex interplay between astrocytes, oligodendrocytes and neurons is only beginning to be understood. In fact, astrocytes have recently been shown to contribute substantial amounts of lipid to oligodendrocytes during myelination [11]. Our findings suggest that oligodendrocytes, that synthesize and preserve a highly lipid-rich environment in the brain, are more involved in osmolyte and energy homeostasis than previously anticipated.
Peripheral ASPA is implicated in juvenile survival
Despite normal survival and the absence of CNS abnormalities, we observed substantial growth retardation in ThyNAT mice that mirrors a peripheral aspect of the pathology observed in AKO and CD. Recent reports have implicated NAA in the energy regulation of peripheral tissues [59, 71]. While not assessed in this study, export of neuron-derived recombinant NAA affecting adipocyte metabolism might be responsible for the observed growth retardation in ThyNAT mice. However, given our investigations were limited to one transgenic line, non-specific position effects affecting physical development cannot be excluded. The virtually normal body weight of Aspa ∆OL mice vs. the diminished body weight of AKO may suggest a role of peripheral ASPA and/or excess NAA for maintaining adipose tissue and lipid turnover. In fact, our findings are in agreement with a report showing that increased NAA, especially in conjunction with reduced ASPA activity, prevents brown adipose tissue cells from expressing mature markers [59].
Disrupted Aspa gene function in AKO and DKO was associated with a biphasic impairment illustrated by a first wave of premature death around four weeks of age. Early phase survivors continued to live until approximately 14 months. These findings match data recently obtained from independent DKO lines [51]. Our data suggest that premature death, observed in DKO and AKO alike, is caused by Aspa deletion, as NKO mice showed normal survival rates. Comparable to DKO and AKO, longevity of Aspa ∆OL mice was reduced indicating that central ASPA dictates long-term survival. These conditional ASPA mutants, lacking central but not peripheral ASPA, showed normal juvenile survival, indicating loss of ASPA expression outside the CNS renders young animals vulnerable to early detrimental events. Furthermore, normal numbers of oligodendrocytes and neurons in DKO underline that impaired survival in this line may have a peripheral origin. AKO and Aspa ∆OL showed substantial loss of both lineage cells. These data show that cell loss is not limited to oligodendrocytes and that NAA depletion prevents cell death, confirming reports in other CD mouse models [44, 69]. Our data do not inform, however, if the loss of one cell lineage precedes demise of the other. The results of our comprehensive phenotypic characterization of NAA-mutant mouse lines are summarized in Table S1. These data have important therapeutic implications as they indicate that even peripheral enzyme replacement therapies could directly benefit CD patients particularly during early stages of the disease.
Aspartoacylase expression in peripheral organs ameliorates Canavan disease
Aspa ∆OL mice, that lack Aspa expression exclusively in oligodendrocytes, were devoid of ASPA-immunoreactivity in the entire CNS, while Aspa expression in peripheral organs was normal. Onset of CD histopathology and behavioural deficiencies were significantly delayed. Despite the fact that Nat8l production was unaltered in these animals, NAA levels in the CNS were markedly lower than in AKO mice. These data indicate that peripheral ASPA functions as a metabolic sink for excess NAA draining off from the CNS. While immunological impairments and decreased macrophage survival have recently been reported in ASPA-null mice [3, 28], the CD pathology is virtually exclusively confined to the CNS. Consequently efforts for development of an enzyme replacement therapy focussed on development of ASPA modification that would allow the recombinant enzyme to cross the blood–brain barrier [58].
Post-symptomatic oligodendroglial ASPA gene therapy reverts Canavan disease pathology
CD is a bona fide target for gene therapy as it is a devastating, fatal childhood disorder and there is no treatment available. In the mouse, a naturally short-lived mammalian species, systemic AAV-mediated ASPA delivery has recently shown great therapeutic success [2, 30]. Nevertheless, immunogenicity and limitations in production continue to impede systemic AAV delivery for treatment of CNS disorders in humans [42]. The CNS is widely recognized as an immune privileged organ as it largely lacks a potent innate immune response [20]. Importantly, direct intracranial AAV-mediated ASPA delivery to CD patients has already proven safe in the clinic [48].
We demonstrated that in the CNS, Aspa is exclusively expressed in oligodendrocytes and that these cells are at the root of CD pathology. We thus hypothesize that complete and long-term therapeutic benefit will be achieved best by restoring ASPA expression in oligodendrocytes, the cell type that naturally expresses the enzyme in the CNS. In line with this hypothesis direct intracranial AAV-ASPA gene delivery to CNS neurons showed limited therapeutic benefit [2, 3, 40], while in a recent study neonatal delivery of a novel AAV serotype that preferably infects oligodendrocytes prevented development of CD in ASPAnur7/nur7 mice, at least until three months of age [25]. In the vast majority of cases, onset of pathology and associated symptoms precede diagnosis. Subsequent gene therapy treatment is likely to be delayed even further. To address this, we aimed to treat AKO mice following manifestation of CD pathology. Based on our previous finding that AAV-mediated transgene expression can be restricted to oligodendrocytes by utilizing oligodendroglial promoters [29, 78, 80] we employed a post-symptomatic Mbp-ASPA gene therapy in AKO mice at P30. Due to its weak immunogenicity, good packaging capacity and infectivity, we utilized cy5, a variant of AAV7 that has been recognized for its potential as clinical gene therapy vector [27]. We achieved oligodendroglial transgenic ASPA expression beyond 9 months of age resulting in normalization of NAA and metabolite concentrations, restoration of CNS pathology present at the time point of injection and neurological amelioration. While gene therapy worked best in young CD patients [48], our preclinical data suggest the presence of a therapeutic window that may be relevant for future clinical studies. However, our post-symptomatic intracranial injection improved, but did not rescue the relative lag of body weight in AKO indicating a peripheral contribution to this phenotype in the CD pathology. The good overall therapeutic outcome would likely to be improved by combining intraparenchymal injections with intrathecal and intracerebroventricular delivery to improve therapeutic ASPA expression in the dorsal brainstem and spinal cord. As the brainstem is involved in the control of vital body functions, direct injection into the brainstem was avoided in this study. Both the intrathecal and intracerebroventricular delivery routes are already successfully performed in clinical trials and well tolerated. In addition, lowering NAA burden through peripheral enzyme replacement therapy, Nat8l knockdown, or via creating an intracranial metabolic sink by ectopic astroglial ASPA expression as proposed by the Gao lab should be considered as a complementary therapeutic strategy [31]. Taken together, this study advances the knowledge around the role of NAA in health and disease and uncovers a feasible roadmap to the successful treatment of CD and other myelin disorders.
Abbreviations
- AAV:
-
Adeno-associated virus
- ASPA:
-
Aspartoacylase
- CD:
-
Canavan disease
- NAT8L:
-
N-Acetyltransferase 8-like
- NAA:
-
N-Acetylaspartate
- NAAG:
-
N-Acetylaspartylglutamate
- AKO:
-
Aspa knockout
- NKO:
-
Nat8l knockout
- DKO:
-
Nat8l/Aspa double-knockout
- ThyNAT:
-
Thy-Nat8l transgenic mice
- Aspa ∆OL :
-
Conditional oligodendrocyte-specific Aspa knockout
- N+/−AKO:
-
Aspa knockout with one deleted Nat8l allele
- AKO-GT:
-
ASPA knockout subjected to gene therapy with AAV-ASPA
- AKO-GFP:
-
ASPA knockout treated with AAV-GFP control vector
References
Amaral AI, Hadera MG, Kotter M, Sonnewald U (2017) Oligodendrocytes do not export NAA-derived aspartate in vitro. Neurochem Res 42:827–837. https://doi.org/10.1007/s11064-016-1985-y
Ahmed SS, Li H, Cao C, Sikoglu EM, Denninger AR, Su Q, Eaton S, Liso Navarro AA, Xie J, Szucs S et al (2013) A single intravenous rAAV injection as late as P20 achieves efficacious and sustained CNS gene therapy in Canavan mice. Mol Ther 21:2136–2147. https://doi.org/10.1038/mt.2013.138
Ahmed SS, Schattgen SA, Frakes AE, Sikoglu EM, Su Q, Li J, Hampton TG, Denninger AR, Kirschner DA, Kaspar B et al (2016) raav gene therapy in a Canavan’s disease mouse model reveals immune impairments and an extended pathology beyond the central nervous system. Mol Ther 24:1030–1041. https://doi.org/10.1038/mt.2016.68
Appu AP, Moffett JR, Arun P, Moran S, Nambiar V, Krishnan JKS, Puthillathu N, Namboodiri AMA (2017) Increasing N-acetylaspartate in the brain during postnatal myelination does not cause the CNS pathologies of Canavan disease. Front Mol Neurosci 10:161. https://doi.org/10.3389/fnmol.2017.00161
Banerjee R, Vitvitsky V, Garg SK (2008) The undertow of sulfur metabolism on glutamatergic neurotransmission. Trends Biochem Sci 33:413–419. https://doi.org/10.1016/j.tibs.2008.06.006
Baslow MH (2002) Evidence supporting a role for N-acetyl-l-aspartate as a molecular water pump in myelinated neurons in the central nervous system. An analytical review. Neurochem Int 40:295–300
Becker I, Lodder J, Gieselmann V, Eckhardt M (2010) Molecular characterization of N-acetylaspartylglutamate synthetase. J Biol Chem 285:29156–29164. https://doi.org/10.1074/jbc.M110.111765
Bothwell JH, Rae C, Dixon RM, Styles P, Bhakoo KK (2001) Hypo-osmotic swelling-activated release of organic osmolytes in brain slices: implications for brain oedema in vivo. J Neurochem 77:1632–1640
Burri R, Steffen C, Herschkowitz N (1991) N-acetyl-l-aspartate is a major source of acetyl groups for lipid synthesis during rat brain development. Dev Neurosci 13:403–411
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA et al (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278. https://doi.org/10.1523/JNEUROSCI.4178-07.2008
Camargo N, Goudriaan A, van Deijk AF, Otte WM, Brouwers JF, Lodder H, Gutmann DH, Nave KA, Dijkhuizen RM, Mansvelder HD et al (2017) Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol 15:e1002605. https://doi.org/10.1371/journal.pbio.1002605
Caroni P (1997) Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods 71:3–9
Chakraborty G, Mekala P, Yahya D, Wu G, Ledeen RW (2001) Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: evidence for myelin-associated aspartoacylase. J Neurochem 78:736–745
Chang L, Munsaka SM, Kraft-Terry S, Ernst T (2013) Magnetic resonance spectroscopy to assess neuroinflammation and neuropathic pain. J Neuroimmune Pharmacol 8:576–593. https://doi.org/10.1007/s11481-013-9460-x
Chen CD, Sloane JA, Li H, Aytan N, Giannaris EL, Zeldich E, Hinman JD, Dedeoglu A, Rosene DL, Bansal R et al (2013) The antiaging protein Klotho enhances oligodendrocyte maturation and myelination of the CNS. J Neurosci 33:1927–1939. https://doi.org/10.1523/jneurosci.2080-12.2013
Chesler M (2003) Regulation and modulation of pH in the brain. Physiol Rev 83:1183–1221. https://doi.org/10.1152/physrev.00010.2003
D’Adamo AF Jr, Yatsu FM (1966) Acetate metabolism in the nervous system. N-acetyl-l-aspartic acid and the biosynthesis of brain lipids. J Neurochem 13:961–965
Davies SE, Gotoh M, Richards DA, Obrenovitch TP (1998) Hypoosmolarity induces an increase of extracellular N-acetylaspartate concentration in the rat striatum. Neurochem Res 23:1021–1025
During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X, Bland RJ, Klugmann M, Banks WA, Drucker DJ et al (2003) Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med 9:1173–1179
Engelhardt B, Vajkoczy P, Weller RO (2017) The movers and shapers in immune privilege of the CNS. Nat Immunol 18:123–131. https://doi.org/10.1038/ni.3666
Estevez AY, O’Regan MH, Song D, Phillis JW (1999) Hyposmotically induced amino acid release from the rat cerebral cortex: role of phospholipases and protein kinases. Brain Res 844:1–9
Fedorov A, Beichel R, Kalpathy-Cramer J, Finet J, Fillion-Robin JC, Pujol S, Bauer C, Jennings D, Fennessy F, Sonka M et al (2012) 3D Slicer as an image computing platform for the quantitative imaging network. Magn Reson Imaging 30:1323–1341. https://doi.org/10.1016/j.mri.2012.05.001
Fisher SK, Novak JE, Agranoff BW (2002) Inositol and higher inositol phosphates in neural tissues: homeostasis, metabolism and functional significance. J Neurochem 82:736–754
Francis JS, Strande L, Markov V, Leone P (2012) Aspartoacylase supports oxidative energy metabolism during myelination. J Cereb Blood Flow Metab 32:1725–1736. https://doi.org/10.1038/jcbfm.2012.66
Francis JS, Wojtas I, Markov V, Gray SJ, McCown TJ, Samulski RJ, Bilaniuk LT, Wang DJ, De Vivo DC, Janson CG et al (2016) N-acetylaspartate supports the energetic demands of developmental myelination via oligodendroglial aspartoacylase. Neurobiol Dis 96:323–334. https://doi.org/10.1016/j.nbd.2016.10.001
Frohlich D, Suchowerska AK, Spencer ZH, von Jonquieres G, Klugmann CB, Bongers A, Delerue F, Stefen H, Ittner LM, Fath T et al (2017) In vivo characterization of the aspartyl-tRNA synthetase DARS: homing in on the leukodystrophy HBSL. Neurobiol Dis 97:24–35. https://doi.org/10.1016/j.nbd.2016.10.008
Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM (2002) Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA 99:11854–11859
Gautier EL, Ivanov S, Williams JW, Huang SC, Marcelin G, Fairfax K, Wang PL, Francis JS, Leone P, Wilson DB et al (2014) Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J Exp Med 211:1525–1531. https://doi.org/10.1084/jem.20140570
Georgiou E, Sidiropoulou K, Richter J, Papaneophytou C, Sargiannidou I, Kagiava A, von Jonquieres G, Christodoulou C, Klugmann M, Kleopa KA (2017) Gene therapy targeting oligodendrocytes provides therapeutic benefit in a leukodystrophy model. Brain 140:599–616. https://doi.org/10.1093/brain/aww351
Gessler DJ, Gao G (2016) Gene therapy for the treatment of neurological disorders: metabolic disorders. Methods Mol Biol 1382:429–465. https://doi.org/10.1007/978-1-4939-3271-9_30
Gessler DJ, Li D, Xu H, Su Q, Sanmiguel J, Tuncer S, Moore C, King J, Matalon R, Gao G (2017) Redirecting N-acetylaspartate metabolism in the central nervous system normalizes myelination and rescues Canavan disease. JCI Insight 2:e90807. https://doi.org/10.1172/jci.insight.90807
Guo F, Bannerman P, Mills Ko E, Miers L, Xu J, Burns T, Li S, Freeman E, McDonough JA, Pleasure D (2015) Ablating N-acetylaspartate prevents leukodystrophy in a Canavan disease model. Ann Neurol 77:884–888. https://doi.org/10.1002/ana.24392
Hao S, Tang B, Wu Z, Ure K, Sun Y, Tao H, Gao Y, Patel AJ, Curry DJ, Samaco RC et al (2015) Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice. Nature 526:430–434. https://doi.org/10.1038/nature15694
Heilig CW, Stromski ME, Blumenfeld JD, Lee JP, Gullans SR (1989) Characterization of the major brain osmolytes that accumulate in salt-loaded rats. Am J Physiol 257:F1108–F1116
Hejazi L, Wong JW, Cheng D, Proschogo N, Ebrahimi D, Garner B, Don AS (2011) Mass and relative elution time profiling: two-dimensional analysis of sphingolipids in Alzheimer’s disease brains. Biochem J 438:165–175. https://doi.org/10.1042/BJ20110566
Ittner LM, Gotz J (2007) Pronuclear injection for the production of transgenic mice. Nat Protoc 2:1206–1215. https://doi.org/10.1038/nprot.2007.145
Karlsson RM, Tanaka K, Saksida LM, Bussey TJ, Heilig M, Holmes A (2009) Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology 34:1578–1589. https://doi.org/10.1038/npp.2008.215
Kaul R, Gao GP, Balamurugan K, Matalon R (1993) Cloning of the human aspartoacylase cDNA and a common missense mutation in Canavan disease. Nat Genet 5:118–123
Ke YD, van Hummel A, Stevens CH, Gladbach A, Ippati S, Bi M, Lee WS, Kruger S, van der Hoven J, Volkerling A et al (2015) Short-term suppression of A315T mutant human TDP-43 expression improves functional deficits in a novel inducible transgenic mouse model of FTLD-TDP and ALS. Acta Neuropathol 130:661–678. https://doi.org/10.1007/s00401-015-1486-0
Klugmann M, Leichtlein CB, Symes CW, Serikawa T, Young D, During MJ (2005) Restoration of aspartoacylase activity in CNS neurons does not ameliorate motor deficits and demyelination in a model of Canavan disease. Mol Ther 11:745–753
Koch M (1999) The neurobiology of startle. Prog Neurobiol 59:107–128
Kotterman MA, Schaffer DV (2014) Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15:445–451. https://doi.org/10.1038/nrg3742
Kubrusly RC, de Mello MC, de Mello FG (1998) Aspartate as a selective NMDA receptor agonist in cultured cells from the avian retina. Neurochem Int 32:47–52
Kumar S, Biancotti JC, Matalon R, de Vellis J (2009) Lack of aspartoacylase activity disrupts survival and differentiation of neural progenitors and oligodendrocytes in a mouse model of Canavan disease. J Neurosci Res 87:3415–3427
Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33:366–374
Le Belle JE, Harris NG, Williams SR, Bhakoo KK (2002) A comparison of cell and tissue extraction techniques using high-resolution 1H-NMR spectroscopy. NMR Biomed 15:37–44
Lehman DM, Harrison JM (2002) Flash visual evoked potentials in the hypomyelinated mutant mouse shiverer. Doc Ophthalmol 104:83–95
Leone P, Shera D, McPhee SW, Francis JS, Kolodny EH, Bilaniuk LT, Wang DJ, Assadi M, Goldfarb O, Goldman HW et al (2012) Long-term follow-up after gene therapy for canavan disease. Sci Trans Med 4:165ra163. https://doi.org/10.1126/scitranslmed.3003454
Madhavarao CN, Arun P, Moffett JR, Szucs S, Surendran S, Matalon R, Garbern J, Hristova D, Johnson A, Jiang W et al (2005) Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan’s disease. Proc Natl Acad Sci USA 102:5221–5226. https://doi.org/10.1073/pnas.0409184102
Madhavarao CN, Moffett JR, Moore RA, Viola RE, Namboodiri MA, Jacobowitz DM (2004) Immunohistochemical localization of aspartoacylase in the rat central nervous system. J Comp Neurol 472:318–329
Maier H, Wang-Eckhardt L, Hartmann D, Gieselmann V, Eckhardt M (2015) N-Acetylaspartate synthase deficiency corrects the myelin phenotype in a Canavan disease mouse model but does not affect survival time. J Neurosci 35:14501–14516. https://doi.org/10.1523/JNEUROSCI.1056-15.2015
Martin E, Capone A, Schneider J, Hennig J, Thiel T (2001) Absence of N-acetylaspartate in the human brain: impact on neurospectroscopy? Ann Neurol 49:518–521
Mehta V, Namboodiri MA (1995) N-Acetylaspartate as an acetyl source in the nervous system. Brain Res Mol Brain Res 31:151–157
Mersmann N, Tkachev D, Jelinek R, Roth PT, Mobius W, Ruhwedel T, Ruhle S, Weber-Fahr W, Sartorius A, Klugmann M (2011) Aspartoacylase-LacZ Knockin Mice: an engineered model of Canavan disease. PLoS ONE 6:e20336. https://doi.org/10.1371/journal.pone.0020336PONE-D-11-01701
Moffett JR, Arun P, Ariyannur PS, Garbern JY, Jacobowitz DM, Namboodiri AM (2011) Extensive aspartoacylase expression in the rat central nervous system. Glia. https://doi.org/10.1002/glia.21186
Moffett JR, Ross B, Arun P, Madhavarao CN, Namboodiri AM (2007) N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol 81:89–131
Pessentheiner AR, Pelzmann HJ, Walenta E, Schweiger M, Groschner LN, Graier WF, Kolb D, Uno K, Miyazaki T, Nitta A et al (2013) NAT8L (N-acetyltransferase 8-like) accelerates lipid turnover and increases energy expenditure in brown adipocytes. J Biol Chem 288:36040–36051. https://doi.org/10.1074/jbc.M113.491324
Poddar NK, Zano S, Natarajan R, Yamamoto B, Viola RE (2014) Enhanced brain distribution of modified aspartoacylase. Mol Genet Metab 113:219–224. https://doi.org/10.1016/j.ymgme.2014.07.002
Prokesch A, Pelzmann HJ, Pessentheiner AR, Huber K, Madreiter-Sokolowski CT, Drougard A, Schittmayer M, Kolb D, Magnes C, Trausinger G et al (2016) N-Acetylaspartate catabolism determines cytosolic acetyl-CoA levels and histone acetylation in brown adipocytes. Sci Rep 6:23723. https://doi.org/10.1038/srep23723
Rae CD (2014) A guide to the metabolic pathways and function of metabolites observed in human brain 1H magnetic resonance spectra. Neurochem Res 39:1–36. https://doi.org/10.1007/s11064-013-1199-5
Rae CD, Davidson JE, Maher AD, Rowlands BD, Kashem MA, Nasrallah FA, Rallapalli SK, Cook JM, Balcar VJ (2014) Ethanol, not detectably metabolized in brain, significantly reduces brain metabolism, probably via action at specific GABA(A) receptors and has measureable metabolic effects at very low concentrations. J Neurochem 129:304–314. https://doi.org/10.1111/jnc.12634
Rigdon GC (1990) Differential effects of apomorphine on prepulse inhibition of acoustic startle reflex in two rat strains. Psychopharmacology 102:419–421
Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM (2000) High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet 25:139–140
Sager TN, Fink-Jensen A, Hansen AJ (1997) Transient elevation of interstitial N-acetylaspartate in reversible global brain ischemia. J Neurochem 68:675–682
Schneider M, Spanagel R, Zhang SJ, Bading H, Klugmann M (2007) Adeno-associated virus (AAV)-mediated suppression of Ca2+/calmodulin kinase IV activity in the nucleus accumbens modulates emotional behaviour in mice. BMC Neurosci 8:105
Schoenfeld R, Wong A, Silva J, Li M, Itoh A, Horiuchi M, Itoh T, Pleasure D, Cortopassi G (2010) Oligodendroglial differentiation induces mitochondrial genes and inhibition of mitochondrial function represses oligodendroglial differentiation. Mitochondrion 10:143–150. https://doi.org/10.1016/j.mito.2009.12.141
Simonato M, Bennett J, Boulis NM, Castro MG, Fink DJ, Goins WF, Gray SJ, Lowenstein PR, Vandenberghe LH, Wilson TJ et al (2013) Progress in gene therapy for neurological disorders. Nat Rev Neurol 9:277–291. https://doi.org/10.1038/nrneurol.2013.56
Singhal NK, Huang H, Li S, Clements R, Gadd J, Daniels A, Kooijman EE, Bannerman P, Burns T, Guo F et al (2016) The neuronal metabolite NAA regulates histone H3 methylation in oligodendrocytes and myelin lipid composition. Exp Brain Res. https://doi.org/10.1007/s00221-016-4789-z
Sohn J, Bannerman P, Guo F, Burns T, Miers L, Croteau C, Singhal NK, McDonough JA, Pleasure D (2017) Suppressing N-acetyl-l-aspartate synthesis prevents loss of neurons in a murine model of Canavan leukodystrophy. J Neurosci 37:413–421. https://doi.org/10.1523/jneurosci.2013-16.2017
Strain GM, Tedford BL (1993) Flash and pattern reversal visual evoked potentials in C57BL/6 J and B6CBAF1/J mice. Brain Res Bull 32:57–63
Surendran S, Matalon R, Tyring SK (2006) Upregulation of aspartoacylase activity in the duodenum of obesity induced diabetes mouse: implications on diabetic neuropathy. Biochem Biophys Res Commun 11:11
Taylor DL, Davies SE, Obrenovitch TP, Doheny MH, Patsalos PN, Clark JB, Symon L (1995) Investigation into the role of N-acetylaspartate in cerebral osmoregulation. J Neurochem 65:275–281
Toriumi K, Kondo M, Nagai T, Hashimoto R, Ohi K, Song Z, Tanaka J, Mouri A, Koseki T, Yamamori H et al (2014) Deletion of SHATI/NAT8L increases dopamine D1 receptor on the cell surface in the nucleus accumbens, accelerating methamphetamine dependence. Int J Neuropsychopharmacol 17:443–453. https://doi.org/10.1017/S1461145713001302
Tranberg M, Abbas AK, Sandberg M (2007) In vitro studies on the putative function of N-acetylaspartate as an osmoregulator. Neurochem Res 32:1248–1255. https://doi.org/10.1007/s11064-007-9300-6
Valsamis B, Schmid S (2011) Habituation and prepulse inhibition of acoustic startle in rodents. J Vis Exp. https://doi.org/10.3791/3446
Verbalis JG (2006) Control of brain volume during hypoosmolality and hyperosmolality. Adv Exp Med Biol 576:113–129. https://doi.org/10.1007/0-387-30172-0_8 (discussion 361–113)
Videen JS, Michaelis T, Pinto P, Ross BD (1995) Human cerebral osmolytes during chronic hyponatremia. A proton magnetic resonance spectroscopy study. J Clin Invest 95:788–793. https://doi.org/10.1172/jci117728
von Jonquieres G, Frohlich D, Klugmann CB, Wen X, Harasta AE, Ramkumar R, Spencer ZH, Housley GD, Klugmann M (2016) Recombinant human myelin-associated glycoprotein promoter drives selective AAV-mediated transgene expression in oligodendrocytes. Front Mol Neurosci 9:13. https://doi.org/10.3389/fnmol.2016.00013
von Jonquieres G, Froud KE, Klugmann CB, Wong AC, Housley GD, Klugmann M (2014) Loss of central auditory processing in a mouse model of Canavan disease. PLoS ONE 9:e97374. https://doi.org/10.1371/journal.pone.0097374
von Jonquieres G, Mersmann N, Klugmann CB, Harasta AE, Lutz B, Teahan O, Housley GD, Frohlich D, Kramer-Albers EM, Klugmann M (2013) Glial promoter selectivity following AAV-delivery to the immature brain. PLoS ONE 8:e65646. https://doi.org/10.1371/journal.pone.0065646
Wang J, Leone P, Wu G, Francis JS, Li H, Jain MR, Serikawa T, Ledeen RW (2009) Myelin lipid abnormalities in the aspartoacylase-deficient tremor rat. Neurochem Res 34:138–148. https://doi.org/10.1007/s11064-008-9726-5
Wibom R, Lasorsa FM, Tohonen V, Barbaro M, Sterky FH, Kucinski T, Naess K, Jonsson M, Pierri CL, Palmieri F et al (2009) AGC1 deficiency associated with global cerebral hypomyelination. N Engl J Med 361:489–495. https://doi.org/10.1056/NEJMoa0900591
Wold S, Antti H, Lindgren F, Ohman J (1998) Orthogonal signal correction of near-infrared spectra. Chemom Intell Lab Syst 44:175–185
Acknowledgements
We thank K.-A. Nave for providing the CNP-Cre mice, and Matthias Eckhardt for the Nat8l cDNA. Nadine Mersmann helped generating floxed ASPA mice. The authors thank the Biological Resources Imaging Laboratory (MWAC, UNSW Sydney) for providing access to instrumentation and operational support, including MRI imaging with National Imaging Facility support. This study utilized the Australian Phenomics Network Histopathology and Organ Pathology Service, University of Melbourne.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This work was supported by grants from the European Commission under the Leukotreat Grant Agreement (FP7-241622), the National Health & Medical Research Council (NH&MRC) Australia (Grant APP1050277 to M.K.), and also by an Australian Research Council (ARC) Future Fellowship (FT100100546 to M.K.), European Leukodystrophies Association (Research Grant: 2015-017I1 to M.K.).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
von Jonquieres, G., Spencer, Z.H.T., Rowlands, B.D. et al. Uncoupling N-acetylaspartate from brain pathology: implications for Canavan disease gene therapy. Acta Neuropathol 135, 95–113 (2018). https://doi.org/10.1007/s00401-017-1784-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1784-9