
Abstract
Sprouty1 (Spry1) is a negative modulator of receptor tyrosine kinase signaling, but its role in cardiomyocyte survival has not been elucidated. The aim of this study was to investigate the potential role of cardiomyocyte Spry1 in cardiac ischemia–reperfusion (I/R) injury. Infarct areas of mouse hearts showed an increase in Spry1 protein expression, which localized to cardiomyocytes. To investigate if cardiomyocyte Spry1 regulates I/R injury, 8-week-old inducible cardiomyocyte Spry1 knockout (Spry1 cKO) mice and control mice were subjected to cardiac I/R injury. Spry1 cKO mice showed reduction in release of cardiac troponin I and reduced infarct size after I/R injury compared to control mice. Similar to Spry1 knockdown in cardiomyocytes in vivo, RNAi-mediated Spry1 silencing in isolated cardiomyocytes improved cardiomyocyte survival following simulated ischemia injury. Mechanistically, Spry1 knockdown induced cardiomyocyte extracellular signal-regulated kinase (ERK) phosphorylation in healthy hearts and isolated cardiomyocytes, and enhanced ERK phosphorylation after I/R injury. Spry1-deficient cardiomyocytes showed better preserved mitochondrial membrane potential following ischemic injury and an increase in levels of phosphorylated ERK and phosphorylated glycogen synthase kinase-3β (GSK-3β) in mitochondria of hypoxic cardiomyocytes. Overexpression of constitutively active GSK-3β abrogated the protective effect of Spry1 knockdown. Moreover, pharmacological inhibition of GSK-3β protected wild-type cardiomyocytes from cell death, but did not further protect Spry1-silenced cardiomyocytes from hypoxia-induced injury. Cardiomyocyte Spry1 knockdown promotes ERK phosphorylation and offers protection from I/R injury. Our findings indicate that Spry1 is an important regulator of cardiomyocyte viability during ischemia–reperfusion injury.
Similar content being viewed by others


Introduction
Cardiovascular diseases are the major cause of mortality worldwide with coronary heart disease accounting to more than 40% of these deaths [13]. The standard of care for the prompt treatment of myocardial infarction (MI) is reperfusion therapies, including primary percutaneous coronary intervention and thrombolysis. A complete coronary occlusion lasting less than 20 min results in reversible injury and myocardial stunning [19, 22]. However, even after prolonged occlusion lasting 4–6 h, 30–50% of the area at risk remains viable. Studies utilizing various cardioprotective agents actually suggest that there is a therapeutic window to alleviate cardiac damage up to 24 h after ischemia [17]. Restoration of blood flow to ischemic myocardium limits infarct size and reduces mortality. The return of blood flow, on the other hand, can also result in additional cardiac damage, referred to as reperfusion injury [8]. Different approaches have been applied to activate cardioprotective signaling (e.g., ERKs and Akt) and to alleviate the ischemic injury to the myocardium (for review, see [16, 20, 43]). However, despite successful preclinical studies with various agents, such as insulin-like growth factor-1, cardiotrophin-1, and erythropoietin, none of these strategies have been sufficient to enter the clinics.
Activation of receptor tyrosine kinases by extracellular ligands (i.e., growth factors) induces phosphorylation of the cytoplasmic tail of the receptor and recruitment of adaptor and effector proteins to the receptor. Activated tyrosine kinase receptors signal to Ras via a protein complex formed by son of sevenless (Sos) and growth factor receptor-bound protein 2 (Grb2) to activate signaling via extracellular signal-regulated kinases (ERKs). ERK is inactivated by dephosphorylation by protein phosphatases such as protein phosphatase 2A and by dual-specificity phosphatase (DUSP)6 [41]. The tyrosine kinase receptor signaling to ERK pathway is also regulated by Sprouty (Spry) proteins, some of which prevent the recruitment of the Sos/Grb2 protein complex and, as a consequence, Ras activation is inhibited. There are four mammalian homologs (Spry1–4), of which all but Spry3 are expressed in the heart [35].
While the role of ERK in cardiomyocyte biology is rather well characterized, there is only little information of Spry proteins. Spry1 expression is increased in failing human hearts following therapy with a left ventricular assist device [23]. In an experimental model, cardiomyocyte-specific overexpression of Spry1 is not sufficient to reduce ERK activity or affect baseline cardiac phenotype [6]. However, the role of Spry1 in cardiomyocytes has not been investigated with loss of function approaches and no data exist on the role of cardiomyocyte Spry1 in regulating cardiomyocyte viability.
In this study, we investigated the role of Spry1 in cardiac ischemia–reperfusion (I/R) injury. We found that Spry1 levels are increased in mouse hearts following acute ischemic injury. We show that suppression of Spry1 in cardiomyocytes activates ERK and p38 mitogen-activated protein kinase (p38) signaling, attenuates cardiac troponin I (cTnI) release and reduces infarct size following I/R injury. Studies in isolated cardiomyocytes show that Spry1 knockdown reduces simulated ischemia-induced mitochondrial permeability transition pore opening and reveal a crucial role for glycogen synthase kinase- 3β (GSK-3β) in mediating the protective effect of Spry1 knockdown.
Materials and methods (detailed methods in Online supplemental materials)
Laboratory animals
All mice were of C57BL/6 background. They were maintained on a 12 h dark/light cycle (6 AM–6 PM) and housed in groups of ≤ 5 with unlimited access to water and chow. The experiments were performed with permission by the national Animal Experiment Board of Finland and by local authorities and conducted in accordance with the Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Spryfl/fl mice, that have been previously published [3], have Lox-P sites flanking open reading frame (ORF) in exon 3 of the mouse Spry1 gene The mice, backcrossed for > 7 generations, were crossed with Myh6-Mer-Cre-Mer mice (Jackson laboratories) in the same background and also backcrossed > 7 generations. Mer-Cre-Mer is constitutively expressed but is inactive until treatment with tamoxifen. At 7 weeks of age, the mice were injected with tamoxifen (40 mg/kg i.p., Cat 13258, Cayman Chemicals, Ann Arbor, MI) on 2 consecutive days. Analysis for genomic DNA from heart samples of Spry1fl/fl × Myh6-Mer-Cre-Mer mice showed efficient Cre-lox recombination in the myocardium upon tamoxifen treatment (Online Fig. 1). At 8 weeks of age, cardiomyocyte Spry1 knockout (Spry1 cKO) and control mice were subjected to cardiac I/R injury.
Myocardial I/R model
Cardiac I/R injury was created by ligation of the left anterior descending artery (LAD) and releasing the slip knot after 30 min to allow for reperfusion of the myocardium as previously described in detail [12]. The mice were thereafter monitored for either 6 or 24 h and cardiac function and extent of cardiac injury (release of cTnI and infarct size or apoptosis) were analyzed. After the I/R procedure, mice were killed by cervical dislocation. Sham-operated mice were subjected to the same surgical procedure without ligation of the LAD. All operations were performed under isoflurane anesthesia (VetEquip vaporizer (VetEquip, Livermore, CA, USA), 2% isoflurane (Baxter, Deerfield, IL, USA), with 1 l/min flow of oxygen). Carprofen (Pfizer, New York, NY, USA) 5 mg/kg s.c. (injection volume 0.125–0.15 ml) was administered as pre-operation analgesia and buprenorphine (Orion Pharma, Espoo, Finland) 0.05 mg/kg s.c. (injection volume 0.1–0.3 ml) was administered as operation analgesia. Post-operation dehydration was prevented with s.c. injection of 5% glucose (injection volume 0.5–1.0 ml). All animals were monitored after the surgery and received a dose of buprenorphine (0.05 mg/kg) in the evening of operation day. Another dose of buprenorphine and a dose of carprofen (5 mg/kg) were administered the following morning.
Cardiomyocyte hypoxia model
Neonatal rat ventricular cardiomyocytes (CMs) were subjected to hypoxia on the fourth day after siRNA transfections. For normoxia cells, DMEM (11966-025, Gibco, Dublin, Ireland) supplemented with 10 mM glucose was used. For hypoxia cells, DMEM supplemented with 10 mM deoxyglucose and 1 mM sodium dithionite (Fluka, Buchs, Switzerland) was used. During hypoxia, cells were incubated in C-Chamber (BioSpherix, New York, NY, USA), with oxygen level controlled at 0.1% with ProOx C21 (BioPherix, New York, NY, USA). When needed 3 µM GSK-3β inhibitor SB216763 (1616, Tocris Bioscience, Bristol, United Kingdom) was added to CMs 20 min before hypoxia experiment.
Statistics
Statistical analysis was performed with IBM SPSS Statistics software (IBM, Armonk, NY, USA). Normally distributed data were analyzed with one-way ANOVA followed by Tukey’s post hoc test. When two groups were compared, Student’s t test was used. If normality was not achieved, data were analyzed with Mann–Whitney test, or in case of three or more groups with Kruskal–Wallis followed by Dunn’s post hoc test. Differences were considered statistically significant at the level of P < 0.05. Data are shown as mean ± SD.
Results
Spry1 protein levels are induced in ischemic cardiomyocytes in vivo
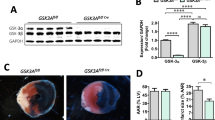
8-Week-old wild-type mice were subjected to experimental MI and protein samples from cardiac tissue were subjected to Western blot analysis. We found that MI induced an increase in levels of phosphorylated ERK and Spry1 protein in the infarcted areas of male myocardium compared to non-ischemic area (septum) that was more clear at 5 h after MI (Fig. 1a). Infarcted tissue of female mouse myocardium also showed induction in phosphorylated ERK and Spry1 expression analyzed 5 h after MI (Fig. 1a). On the other hand, myocardial infarction had no effect on expression of other cardiac Sprouty proteins, Spry2 or Spry4 (Fig. 1a).

Analysis for Spry1 expression in ischemic myocardium. a Wild-type male mice were subjected to myocardial infarction (MI) for 1 and 5 h, and female mice for 5 h. Heart tissues from infarcted area and remote area (septum) were collected. Shown is immunoblot analysis of Spry1, phosphorylated extracellular signal-regulated kinase (p-ERK), Spry2, Spry4 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Quantification of immunoblot analysis of Spry1 and p-ERK. GAPDH was used as a loading control. Data are shown as fold versus septum. N = 4–5; *P < 0.05, **P < 0.01, ***P < 0.001 versus septum. b Spry1fl/fl mice were subjected to ischemia–reperfusion (I/R) injury and heart tissue was collected after 6 h of reperfusion. Shown is the immunofluorescence staining of formalin fixed and paraffin-embedded tissue sections from infarct area for Spry1 and cardiomyocyte marker α-actinin. Scale bar 40 µm
We then investigated for Spry1 expression in resident cardiac cell types. In accordance with previous data [50], we found that Spry1 mRNA is expressed at eight-fold higher levels in fibroblast fraction compared to cardiomyocyte fraction in healthy hearts (Online Fig. 2). To assess if the increase in Spry1 protein expression occurs in cardiomyocytes in ischemic hearts, we subjected 8-week old male Spry1fl/fl mice to I/R injury for 6 h and analyzed for localization of Spry1 expression by immunofluorescence microscopy. Analysis of cardiac sections showed that Spry1 is induced in ischemic areas in hearts of Spry1fl/fl mice and colocalizes with cardiomyocyte marker α-actinin (Fig. 1b).
Cardiomyocyte Spry1 knockdown protects from cardiac I/R injury
To investigate for the role of cardiomyocyte Spry1 in regulation of cardiac ischemic injury, we utilized tamoxifen-inducible Cre-lox technique to delete Spry1 specifically from the cardiomyocytes. Wild-type (WT), Spry1fl/fl and Spry1 cKO mice were subjected to cardiac I/R injury with 30 min of ischemia followed by 24 h of reperfusion. Analysis of cardiac structure and function by echocardiography prior to cardiac ischemia showed no difference between WT, Spry1fl/fl and Spry1 cKO mice (Online Table 1). Analysis for the extent of cardiac injury at 24 h after I/R showed that Spry1 knockdown in cardiomyocytes had no effect on area at risk, but significantly reduced infarct size compared to both WT and Spry1fl/fl mice (Fig. 2a). Analysis of plasma samples showed that Spry1 cKO mice had significantly lower plasma cTnI levels at the 24 h after I/R injury compared to both WT and Spry1fl/fl mice (Fig. 2b). Echocardiography analysis showed significantly better preserved systolic function in Spry1 cKO mice compared to WT mice, while the difference between Spry1 cKO and Spry1fl/fl mice did not reach statistical significance (Table 1). No difference was observed in left ventricular end-diastolic diameter between the groups, but the stroke volume was greater in Spry1 cKO mice compared to Spry1fl/fl or WT mice (Table 1). Spry1 cKO mice also showed a 20% increase in cardiac output compared to control mice (Table 1).

Spry1 cardiomyocyte knockdown protects from ischemia–reperfusion injury in vivo. Wild-type (WT), Spry1fl/fl and Spry1 cardiomyocyte knockout (cKO) mice were subjected to 30 min of cardiac ischemia and 24 h of reperfusion, and subjected to analysis of infarct size, serum troponin I levels and analysis of cardiac function by echocardiography. a Analysis for area at risk (AAR) and infarct size relative to AAR derived from triphenyltetrazolium chloride (TTC) stainings. N = 9–13 per group. b Analysis for troponin I levels from serum samples. N = 9–13 per group. c Spry1fl/fl and Spry1 cKO mice were subjected to 30 min of ischemia and 6 h of reperfusion, and subjected to analysis for serum troponin I levels and apoptosis by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). Shown are also representative images of TUNEL labeling of sections from hearts of Spry1fl/fl and Spry1 cKO mice. Scale bar 40 µm. N = 11–13 per group. *P < 0.05 versus WT mice and #P < 0.05, ##P < 0.01 versus Spry1fl/fl mice
Spry1fl/fl mice and Spry1 cKO mice were then subjected to cardiac I/R injury for 6 h. Analysis of plasma samples showed significantly lower plasma cTnI levels in Spry cKO mice compared to the Spry1fl/fl mice (Fig. 2c). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining of left ventricular (LV) sections showed a significant reduction in the number of apoptotic cells in ischemic areas of Spry1 cKO mouse hearts compared to the Spry1fl/fl mouse hearts (Fig. 2c).
We next investigated if cardiomyocyte Spry1 deletion provides protection from I/R injury in female hearts. 8-Week-old female Spry1fl/fl mice and Spry1 cKO mice were subjected to cardiac I/R injury for 24 h. Analysis for infarct size showed no difference in area at risk, but revealed a significant reduction in infarct size in female Spry1 cKO mice compared to female Spry1fl/fl mice (Online Fig. 3a). Analysis of plasma samples of female mice showed a significant decrease in plasma cTnI levels in Spry cKO mice compared to the Spry1fl/fl mice (Online Fig. 3b).
Spry1 knockdown protects isolated cardiomyocytes from hypoxic injury
We then investigated if depletion of Spry1 in isolated cardiomyocytes offers protection from hypoxic injury. Spry1 RNAi in adult rat primary cardiomyocytes proved fruitless, since Spry1 mRNA expression was downregulated by 80–90% in non-treated healthy cardiomyocytes cultured for 2–4 days (Online Fig. 4). We then utilized RNAi in neonatal rat ventricular cardiomyocytes (CMs) which preserve their Spry1 expression in culture. Treatment of CMs with Spry1 siRNA led to 76% decrease in Spry1 mRNA and ~ 60% decrease in Spry1 protein levels (Fig. 3a). To investigate the effect of Spry1 RNAi on cardiomyocyte survival, Spry1 siRNA-treated CMs were subjected to simulated ischemia injury or cardiotoxic injury by doxorubicin treatment. Spry1-deficient cells showed markedly better survival [as assessed by release of adenylate kinase (AK) from ruptured cells] following 4 h of simulated ischemia (Fig. 3b). Spry1 knockdown, however, did not affect cardiomyocyte survival following treatment with 3 µM or 10 µM doxorubicin (Fig. 3c).

Spry1 knockdown in isolated cardiomyocytes protects from hypoxia–reperfusion injury. Neonatal rat ventricular cardiomyocytes (CMs) were transfected with Spry1 siRNA (100 nM) or control siRNA (100 nM). a Three days later, RNA samples were collected from CMs and analyzed by qPCR. Shown is the relative expression of Spry1 mRNA in Spry1 RNAi treated and control RNAi treated CMs. Results were normalized to expression of 18S ribosomal RNA (18S). N = 3 for each group. Four days later, protein samples were collected from CMs and analyzed by western blotting. Shown is immunoblot analysis for Spry1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and quantification of immunoblot analysis of Spry1. GAPDH was used as a loading control. Data are shown as fold versus control RNAi. N = 3–6. b RNAi-treated CMs were subjected to hypoxia. Shown is the analysis for released adenylate kinase (AK) from damaged CMs after 4 h of hypoxia. N = 4 for each group. c RNAi-treated CMs were treated with 3 μM and 10 μM doxorubicin for 24 h. Shown is the analysis for AK release from damaged CMs. N = 3 for each group. ***P < 0.001 versus control RNAi; ###P < 0.001 versus normoxia control RNAi and §§P < 0.01 versus control RNAi in hypoxia
Spry1 regulates ERK and p38 in cardiomyocytes
As Spry proteins have been shown to inhibit fibroblast growth factor induced Raf-ERK pathway activation [15], we assessed the effect of cardiomyocyte Spry1 knockdown on ERK activity. Western blot analysis for LV samples from healthy Spry1 cKO mice showed an increase in ERK phosphorylation in the myocardium compared to control Spry1fl/fl mice (Fig. 4a, Online Fig. 5a). Analysis of ERK phosphorylation at 6 h after I/R injury showed that ERK phosphorylation remained elevated in ischemic myocardium of Spry1 cKO compared to ischemic areas of Spry1fl/fl mouse hearts (Fig. 4a, Online Fig. 5a). In agreement with data from hearts in vivo, Spry1 knockdown in isolated cardiomyocytes increased ERK phosphorylation (Fig. 4b, Online Fig. 5b). Immunofluorescence analysis of cardiac sections showed that ERK is mainly phosphorylated in the vascular wall in hearts of sham-operated Spry1fl/fl mice (Fig. 4c), whereas Spry1 cKO mice show ERK phosphorylation in cardiomyocytes (Fig. 4c). Immunofluorescence analysis after 6 h of I/R injury showed that ERK phosphorylation in cardiomyocytes is increased in both genotypes in ischemic areas, but Spry1 cKO cardiomyocytes show more intense staining (Fig. 4c). Spry1 thus negatively regulates ERK phosphorylation in cardiomyocytes at both baseline and following I/R injury.

Spry1 regulates ERK and p38 in cardiomyocytes. Spry1fl/fl and Spry1 cardiomyocyte knockout (cKO) mice were subjected to sham operation or to 30 min of ischemia (I/R), and heart tissue was collected after 6 h of reperfusion. a Immunoblot analysis of phosphorylated extracellular signal-regulated kinase (p-ERK) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in sham and I/R operated Spry1fl/fl and Spry1 cKO hearts. b Neonatal rat ventricular cardiomyocytes were transfected with Spry1 siRNA (100 nM) or control siRNA (100 nM). Four days later, protein samples were collected. Shown is the immunoblot analysis for Spry1, p-ERK and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). c Immunofluorescence analysis for p-ERK and F-actin marker Phalloidin from control hearts and infarct areas from hearts subjected to 6 h of I/R injury. Scale bar 40 µm. d Immunoblot analysis of phosphorylated p38 (p-p38) and GAPDH in sham and I/R operated Spry1fl/fl and Spry1 cKO hearts. e Immunoblot analysis for Spry1, p-p38 and GAPDH from protein samples of neonatal rat ventricular cardiomyocytes transfected with Spry1 siRNA (100 nM) or control siRNA (100 nM)
Analysis of other central signaling pathways in cardiomyocytes showed that phosphorylation of p38 was increased in hearts of healthy Spry1 cKO mice compared to Spryfl/fl mice (Fig. 4d, Online Fig. 5c). However, no difference was observed in p38 phosphorylation in ischemic tissues of Spry1fl/fl and Spry1 cKO mice at 6 h after I/R injury (Fig. 4d, Online Fig. 5c). Analysis of other central cell survival pathways [phosphoinositide 3-kinase (PI3K)-Akt, Signal transducer and activator of transcription 3 (STAT3), c-Jun N-terminal kinase] showed no difference between hearts of Spryfl/fl and Spry1 cKO mice either prior to ischemia or after the ischemia. In accordance with in vivo findings, Spry1 knockdown in isolated neonatal cardiomyocytes showed increased p38 phosphorylation (Fig. 4e, Online Fig. 5d). Spry1 thus regulates p38 phosphorylation in non-injured cardiomyocytes.
Spry1 knockdown inhibits mitochondrial GSK-3β to protect from hypoxic injury
Opening of the mitochondrial permeability transition pore (mPTP) at reperfusion is a critical determinant of I/R injury and inhibition of mPTP opening has been shown to reduce MI size and preserve cardiac function both in preclinical and clinical studies [5, 26]. To investigate if Spry1 regulates mPTP opening, we first investigated for Spry1 localization in injured cardiomyocytes. Analysis of LV sections from Spry1fl/fl mice subjected to cardiac I/R injury for 6 h showed co-staining of Spry1 and mitochondrial marker heat shock protein 60 (HSP60) in cardiomyocytes (Fig. 5a). To determine if Spry1 regulates mitochondrial membrane potential (ΔΨm), Spry1 and control siRNA treated cardiomyocytes were subjected to 4 h of hypoxia and ΔΨm was measured with JC-1. We found that Spry1-deficient cardiomyocytes have significantly better preserved mitochondrial membrane potential compared to control cardiomyocytes (Fig. 5b). Similarly, analysis of mPTP opening by calcein cobalt assay showed better preserved mitochondrial membrane integrity in Spry1-deficient cardiomyocytes (Fig. 5b). Inhibition of GSK-3β has been shown to reduce mitochondrial membrane permeability transition and cell death, and ERK and p38 signaling pathways have both been shown to phosphorylate and inhibit GSK-3β [10, 25, 34, 49]. We then performed western blot analyses of pooled mitochondrial fractions from cardiomyocytes subjected to hypoxia and found that the levels of phosphorylated ERK and phosphorylated (inactive) GSK-3β were increased in mitochondria of Spry1-deficient cardiomyocytes compared to control siRNA transfected cardiomyocytes (Fig. 5c). To assess for possible role of GSK-3β in Spry1-mediated response, we transduced isolated cardiomyocytes with adenoviruses encoding for wild-type GSK-3β, GSK-3β harboring a Ser9Ala mutation and GSK-3β harboring Ser9Ala and Ser389Ala mutations. Phosphorylation of GSK-3β at Ser9 or at Ser389 has previously been shown to inactivate the kinase [48, 49]. We found that the forced activation of GSK-3β has no effect on hypoxia-induced cell death in control siRNA-treated cardiomyocytes (Fig. 5d). However, overexpression of GSK-3β (Ser9Ala) partially reverses the protective effect of Spry1 knockdown in hypoxic cardiomyocytes and overexpression of GSK-3β (Ser9Ala/Ser389Ala) completely abolishes the protective effect of Spry1 knockdown (Fig. 5d). Finally, we investigated if control and Spry1 siRNA-treated cardiomyocytes were sensitive to GSK-3β inhibition during hypoxia. We found that the treatment of control cardiomyocytes with SB216763 (3 μM) significantly reduces hypoxia-induced cardiomyocyte death (Fig. 5e). However, pharmacological inhibition of GSK-3β does not further protect Spry1-deficient cardiomyocytes from hypoxia-induced injury (Fig. 5e). These data thus indicate a crucial role for GSK-3β in regulating the protective response to Spry1 knockdown.

Glycogen synthase kinase-3β mediates the protective effect of Spry1 knockdown. a Spry1fl/fl mice were subjected to 30 min of ischemia and heart tissue was collected after 6 h of reperfusion. Shown is the immunofluorescence staining of frozen sections from infarct area for Spry1 and mitochondrial marker heat shock protein 60 (HSP60). Scale bar 40 µm. b Neonatal rat ventricular cardiomyocytes (CMs) were transfected with Spry1 siRNA (100 nM) or control siRNA (100 nM), and 4 days later subjected to hypoxia for 4 h. Shown is the analysis for mitochondrial membrane potential expressed as ratio of JC-1 aggregate versus JC-1 monomer and detection of mitochondrial permeability transition pore opening using calcein cobalt assay expressed as relative Calcein-AM fluorescence (%). N = 4–8; **P < 0.01, ***P < 0.001 versus normoxia control RNAi, #P < 0.05, ###P < 0.001 versus hypoxia control RNAi. c RNAi treated CMs were subjected to hypoxia for 4 h and mitochondrial protein samples were collected and analyzed by western blotting for phosphorylated extracellular signal-regulated kinase (p-ERK), phosphorylated glycogen synthase kinase-3β (p-GSK-3β) and HSP60. d RNAi-treated CMs were transduced with adenoviruses expressing LacZ, wild-type (WT) GSK-3β, Ser9Ala GSK-3β or Ser9Ala/Ser389Ala double mutant GSK-3β, and subjected to hypoxia for 4 h. Shown is the analysis for released adenylate kinase (AK) from damaged CMs after hypoxia. N = 4 for each group; ***P < 0.001 versus normoxia control RNAi, ##P < 0.01 versus hypoxia control RNAi and §P < 0.05, §§P < 0.01 versus hypoxia Spry1 RNAi. e RNAi-treated CMs were treated with GSK-3β inhibitor SB216763 (3 μM) for 20 min and subjected to hypoxia for 4 h. Shown is the analysis for released AK from damaged CMs after hypoxia. N = 4 each group; ***P < 0.001 versus normoxia control RNAi, ###P < 0.001 versus hypoxia control RNAi
Discussion
Despite an increasing number of patients suffering from ischemic heart disease, there are no pharmacological therapies in clinical use for treatment of cardiac I/R injury. Activation of RISK and Safe pathways (i.e., ERK, STAT and PI3K-Akt) by various growth factors and pharmacological agents have shown beneficial effects in attenuating ischemic cardiomyocyte injury in experimental models [18, 20]. However, this has not led to development of novel therapies. One important caveat with these approaches is that even with continuous growth factor stimulation, the activity of pro-survival pathways in cardiomyocytes is usually transient [47]. Growth factor-induced activation of ERK, for example, is abrogated in response to inhibitory feedback signaling to Ras and actually to nearly all ERK cascade components, by downstream kinases as well as by induction of inhibitors of tyrosine kinase signaling, such as Sprouty [27, 28]. In the context of G protein-coupled receptor (GPCR) activation, activated GPCRs are desensitized by phosphorylation by members of the family of G protein-coupled receptor kinases (GRKs) [11].
In the current study, we found that ischemia resulted in accumulation of Spry1 protein in cardiomyocytes in ischemic myocardium, but had no effect on expression of other Spry proteins in the myocardium. We then utilized cardiomyocyte Spry1 knockdown both in vivo and in vitro to investigate the role of Spry1 in cardiac I/R injury. We found that cardiomyocyte Spry1 knockdown in vivo reduced infarct size, reduced troponin I release and reduced the number of TUNEL positive cells in the myocardium following I/R injury. Data from in vitro studies showed that Spry1 knockdown was sufficient to reduce cardiomyocyte death following experimental hypoxia. Analysis of downstream signaling pathways regulated by Spry1 showed that Spry1 knockdown results in activation of ERK and p38 pathways in cardiomyocytes. Data from previous studies indicate that the key mechanism by which Spry proteins modulate cell proliferation and survival is inhibition of ERK pathway [31]. On the other hand, inhibition of Spry proteins in various cell types has been shown to induce robust ERK activation that varies in strength and duration [33]. ERK phosphorylation in the myocardium is induced following experimental ischemia and in human hearts in response to cardioplegia/reperfusion [21, 42]. Interestingly, both induction of ERK signaling and hypoxia have been shown to induce Spry1 expression [1, 40]. The noted increase in Spry1 levels in cardiomyocytes after ischemic injury may thus serve as a feedback mechanism to limit ERK activation. The role of the ERK cascade in regulating cardiomyocyte survival is well characterized. Inhibition of ERKs has been shown to exaggerate I/R-induced apoptosis and, conversely, increased ERK phosphorylation/activation is associated with protection from cardiac I/R injury [7, 29, 51]. However, the downstream mechanisms mediating the protective effects of ERK are not fully understood [42]. We found that Spry1 depletion in mouse cardiomyocytes augmented ERK phosphorylation after I/R injury and, notably, induced ERK activation at baseline, indicating that cardiomyocyte Spry1 regulates ERK in physiological conditions. Spry1 thus appears to be a central regulator of ERK in cardiomyocytes and, differently from ligand-induced ERK activation, Spry1 depletion results in persistent ERK phosphorylation that is preferable for a cardioprotective agent.
In addition to ERKs, we found that depletion of Spry1 in cardiomyocytes in vivo and in vitro resulted in increased p38 phosphorylation. Previously, activation of p38 signaling has only been observed in triple Spry1/Spry2/Spry4 knockouts in mouse embryonic fibroblasts [46]. p38 appears to have a dual role in myocardial I/R injury. p38 is activated rapidly in response to myocardial ischemia [30] and there are data indicating that inhibition of p38 during ischemia and/or reperfusion is cardioprotective [9, 20]. However, a clinical trial with losmapimod, an oral nonisoform selective inhibitor of p38, found no effect on necrosis biomarkers creatine kinase and TnI in patients with non-ST segment elevation acute MI [39]. On the other hand, the activity of p38 is increased during ischemic preconditioning and its inhibition abrogates the cardioprotection [36,37,38, 45]. p38 activation during preconditioning in vivo has actually been shown to attenuate p38 activation during ischemia [44]. In the current study, we found that Spry1 depletion both in vivo and in vitro induced phosphorylation of p38. However, cardiomyocyte Spry1 knockdown had no effect on p38 phosphorylation after I/R injury.
Opening of the mPTP has been shown to play a crucial role in cardiac I/R injury and ischemic postconditioning attenuating the mitochondrial permeability transition [2, 26]. Interestingly, there is evidence from NIH3T3 cells that Spry2 localizes in mitochondrial membranes [32]. In the current study, we found that the induction in Spry1 expression in cardiomyocytes after I/R injury was localized to cardiomyocyte mitochondria and Spry1 inhibition attenuated mitochondrial membrane permeability transition. Furthermore, Spry1 depletion did not protect from doxorubicin-induced cardiomyocyte death, which is triggered by mechanisms other than mitochondrial membrane permeability transition, such as through mitochondrial iron accumulation and topoisomerase-IIβ. While cyclophilin D is the best-characterized regulator of the mPTP, prior studies have also indicated a role for GSK-3β in regulating mPTP opening [14]. Genetic inactivation of GSK-3β has been shown to protect from I/R injury, whereas forced activation of GSK-3β exacerbated myocardial I/R injury [52]. Multiple signaling pathways have been shown to converge at the level of GSK-3β resulting in increased phosphorylation and inhibition of the kinase [4, 24, 25]. ERK has been shown to phosphorylate GSK-3β at the Thr43 residue priming GSK-3β for its subsequent phosphorylation at Ser9, resulting in inactivation of GSK-3β [10]. GSK-3β is also phosphorylated and inactivated at Ser389 by p38 [49], but the significance of this phosphorylation in cardiomyocytes is not known. We found that mitochondrial GSK-3β was Ser9 phosphorylated (i.e., inhibited) in Spry1-deficient cardiomyocytes following hypoxia. We then investigated if GSK-3β played a role in protective response to Spry1 knockdown and found that overexpression of constitutively active GSK-3β (Ser9Ala) modestly attenuated the protective effect of Spry1 knockdown. However, overexpression of GSK-3β also harboring a p38 target site mutation (Ser9Ala/Ser389Ala) fully abrogated the protective effect of Spry1 inhibition. On the other hand, overexpression of active forms of GSK-3β did not increase cell death in control cardiomyocytes subjected to hypoxia. In accord with the actions of active GSK-3β, pharmacological inhibition of GSK-3β did not further protect Spry1-deficient cardiomyocytes from hypoxic injury, while GSK-3β inhibition significantly reduced hypoxia-induced cell death in control cardiomyocytes. These data thus indicate that in the context of I/R injury, mitochondrial GSK-3β is a central downstream target of Spry1. However, given the robust activation of ERK and p38 upon Spry1 depletion, other mechanisms may also play a role in mediating the cardioprotective effect.
In summary, we found that myocardial ischemia promotes Spry1 protein expression in ischemic cardiomyocytes. Our experimental data show that cardiomyocyte Spry1 knockdown enhances ERK phosphorylation and protects from cardiac I/R injury. Data from the studies in isolated cardiomyocytes show that Spry1 knockdown protects from hypoxia-induced mitochondrial permeability transition pore opening and indicates a central role for GSK-3β in mediating the protective effect of Spry1 knockdown. Further studies are needed to evaluate if inhibition of Spry1 offers novel therapeutic strategy to alleviate cardiac ischemia–reperfusion injury.
References
Anderson K, Nordquist KA, Gao X, Hicks KC, Zhai B, Gygi SP, Patel TB (2011) Regulation of cellular levels of Sprouty2 protein by prolyl hydroxylase domain and von Hippel–Lindau proteins. J Biol Chem 286:42027–42036. https://doi.org/10.1074/jbc.M111.303222
Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M (2005) Postconditioning inhibits mitochondrial permeability transition. Circulation 111:194–197. https://doi.org/10.1161/01.CIR.0000151290.04952.3B
Basson MA, Akbulut S, Watson-Johnson J, Simon R, Carroll TJ, Shakya R, Gross I, Martin GR, Lufkin T, McMahon AP, Wilson PD, Costantini FD, Mason IJ, Licht JD (2005) Sprouty1 is a critical regulator of GDNF/RET-mediated kidney induction. Dev Cell 8:229–239. https://doi.org/10.1016/j.devcel.2004.12.004
Beurel E, Grieco SF, Jope RS (2015) Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 148:114–131. https://doi.org/10.1016/j.pharmthera.2014.11.016
Botker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Di Lisa F, Di Sante M, Efentakis P, Femmino S, Garcia-Dorado D, Giricz Z, Ibanez B, Iliodromitis E, Kaludercic N, Kleinbongard P, Neuhauser M, Ovize M, Pagliaro P, Rahbek-Schmidt M, Ruiz-Meana M, Schluter KD, Schulz R, Skyschally A, Wilder C, Yellon DM, Ferdinandy P, Heusch G (2018) Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol 113:39. https://doi.org/10.1007/s00395-018-0696-8
Charles NJ, Huebert RC, Lee S, Adhikari N, Polster S, Rider JE, Braunlin E, Mariash A, Robledo M, Schuweiler D, Hall JL (2010) Targeted Sprouty1 overexpression in cardiac myocytes does not alter myocardial remodeling or function. Mol Cell Biochem 342:57–62. https://doi.org/10.1007/s11010-010-0468-8
Das A, Salloum FN, Xi L, Rao YJ, Kukreja RC (2009) ERK phosphorylation mediates sildenafil-induced myocardial protection against ischemia-reperfusion injury in mice. Am J Physiol Heart Circ Physiol 296:H1236–H1243. https://doi.org/10.1152/ajpheart.00100.2009
Davidson SM, Arjun S, Basalay MV, Bell RM, Bromage DI, Botker HE, Carr RD, Cunningham J, Ghosh AK, Heusch G, Ibanez B, Kleinbongard P, Lecour S, Maddock H, Ovize M, Walker M, Wiart M, Yellon DM (2018) The 10th Biennial Hatter Cardiovascular Institute workshop: cellular protection-evaluating new directions in the setting of myocardial infarction, ischaemic stroke, and cardio-oncology. Basic Res Cardiol 113:43. https://doi.org/10.1007/s00395-018-0704-z
Denise Martin E, De Nicola GF, Marber MS (2012) New therapeutic targets in cardiology: p38 alpha mitogen-activated protein kinase for ischemic heart disease. Circulation 126:357–368. https://doi.org/10.1161/CIRCULATIONAHA.111.071886
Ding Q, Xia W, Liu J-C, Yang J-Y, Lee D-F, Xia J, Bartholomeusz G, Li Y, Pan Y, Li Z, Bargou RC, Qin J, Lai C-C, Tsai F-J, Tsai C-H, Hung M-C (2005) Erk associates with and primes GSK-3β for Its inactivation resulting in upregulation of β-catenin. Mol Cell 19:159–170. https://doi.org/10.1016/j.molcel.2005.06.009
Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG (2004) Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci 27:107–144. https://doi.org/10.1146/annurev.neuro.27.070203.144206
Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL, Koch WJ (2010) A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res 107:1445–1453. https://doi.org/10.1161/CIRCRESAHA.110.223925
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER III, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB (2014) Heart disease and stroke statistics-2014 update: a report from the American Heart Association. Circulation 129:e28–e292. https://doi.org/10.1161/01.cir.0000441139.02102.80
Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M (2008) Inhibition of GSK3β by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation 117:2761–2768. https://doi.org/10.1161/circulationaha.107.755066
Hanafusa H, Torii S, Yasunaga T, Nishida E (2002) Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol 4:850–858. https://doi.org/10.1038/ncb867
Hausenloy DJ, Barrabes JA, Bøtker HE, Davidson SM, Di Lisa F, Downey J, Engstrom T, Ferdinandy P, Carbrera-Fuentes HA, Heusch G, Ibanez B, Iliodromitis EK, Inserte J, Jennings R, Kalia N, Kharbanda R, Lecour S, Marber M, Miura T, Ovize M, Perez-Pinzon MA, Piper HM, Przyklenk K, Schmidt MR, Redington A, Ruiz-Meana M, Vilahur G, Vinten-Johansen J, Yellon DM, Garcia-Dorado D (2016) Ischaemic conditioning and targeting reperfusion injury: a 30 year voyage of discovery. Basic Res Cardiol 111:70. https://doi.org/10.1007/s00395-016-0588-8
Hausenloy DJ, Yellon DM (2013) Myocardial ischemia–reperfusion injury: a neglected therapeutic target. J Clin Invest 123:92–100. https://doi.org/10.1172/JCI62874
Hausenloy DJ, Yellon DM (2006) Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res 70:240–253. https://doi.org/10.1016/j.cardiores.2006.01.017
Heusch G (2016) The coronary circulation as a target of cardioprotection. Circ Res 118:1643–1658. https://doi.org/10.1161/CIRCRESAHA.116.308640
Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699. https://doi.org/10.1161/circresaha.116.305348
Heusch G, Musiolik J, Kottenberg E, Peters J, Jakob H, Thielmann M (2012) STAT5 activation and cardioprotection by remote ischemic preconditioning in humans: short communication. Circ Res 110:111–115. https://doi.org/10.1161/CIRCRESAHA.111.259556
Heyndrickx GR, Millard RW, McRitchie RJ, Maroko PR, Vatner SF (1975) Regional myocardial functional and electrophysiological alterations after brief coronary artery occlusion in conscious dogs. J Clin Invest 56:978–985. https://doi.org/10.1172/JCI108178
Huebert RC, Li Q, Adhikari N, Charles NJ, Han X, Ezzat MK, Grindle S, Park S, Ormaza S, Fermin D, Miller LW, Hall JL (2004) Identification and regulation of Sprouty1, a negative inhibitor of the ERK cascade, in the human heart. Physiol Genom 18:284–289. https://doi.org/10.1152/physiolgenomics.00098.2004
Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ (2004) Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113:1535–1549. https://doi.org/10.1172/JCI19906
Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ (2009) Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res 104:1240–1252. https://doi.org/10.1161/CIRCRESAHA.109.197996
Kwong Jennifer Q, Molkentin Jeffery D (2015) Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab 21:206–214. https://doi.org/10.1016/j.cmet.2014.12.001
Lavoie H, Therrien M (2015) Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 16:281–298. https://doi.org/10.1038/nrm3979
Lemmon MA, Schlessinger J (2010) Cell signaling by receptor-tyrosine kinases. Cell 141:1117–1134. https://doi.org/10.1016/j.cell.2010.06.011
Lips DJ, Bueno OF, Wilkins BJ, Purcell NH, Kaiser RA, Lorenz JN, Voisin L, Saba-El-Leil MK, Meloche S, Pouyssegur J, Pages G, De Windt LJ, Doevendans PA, Molkentin JD (2004) MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation 109:1938–1941. https://doi.org/10.1161/01.CIR.0000127126.73759.23
Ma XL, Kumar S, Gao F, Louden CS, Lopez BL, Christopher TA, Wang C, Lee JC, Feuerstein GZ, Yue TL (1999) Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation 99:1685–1691. https://doi.org/10.1161/circ.99.13.1685
Mason JM, Morrison DJ, Albert Basson M, Licht JD (2006) Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol 16:45–54. https://doi.org/10.1016/j.tcb.2005.11.004
Mason JM, Morrison DJ, Bassit B, Dimri M, Band H, Licht JD, Gross I (2004) Tyrosine phosphorylation of sprouty proteins regulates their ability to inhibit growth factor signaling: a dual feedback loop. Mol Biol Cell 15:2176–2188. https://doi.org/10.1091/mbc.E03-07-0503
Masoumi-Moghaddam S, Amini A, Morris DL (2014) The developing story of Sprouty and cancer. Cancer Metastasis Rev 33:695–720. https://doi.org/10.1007/s10555-014-9497-1
Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR (2006) Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 21:749–760. https://doi.org/10.1016/j.molcel.2006.02.009
Minowada G, Jarvis LA, Chi CL, Neubuser A, Sun X, Hacohen N, Krasnow MA, Martin GR (1999) Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development 126:4465–4475
Mocanu MM, Baxter GF, Yue Y, Critz SD, Yellon DM (2000) The p38 MAPK inhibitor, SB203580, abrogates ischaemic preconditioning in rat heart but timing of administration is critical. Basic Res Cardiol 95:472–478. https://doi.org/10.1007/s003950070023
Nakano A, Baines CP, Kim SO, Pelech SL, Downey JM, Cohen MV, Critz SD (2000) Ischemic preconditioning activates MAPKAPK2 in the isolated rabbit heart: evidence for involvement of p38 MAPK. Circ Res 86:144–151. https://doi.org/10.1161/res.86.2.144
Nakano A, Cohen MV, Critz S, Downey JM (2000) SB 203580, an inhibitor of p38 MAPK, abolishes infarct-limiting effect of ischemic preconditioning in isolated rabbit hearts. Basic Res Cardiol 95:466–471. https://doi.org/10.1007/s003950070022
Newby LK, Marber MS, Melloni C, Sarov-Blat L, Aberle LH, Aylward PE, Cai G, de Winter RJ, Hamm CW, Heitner JF, Kim R, Lerman A, Patel MR, Tanguay JF, Lepore JJ, Al-Khalidi HR, Sprecher DL, Granger CB, Investigators S (2014) Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: a randomised phase 2 trial. Lancet 384:1187–1195. https://doi.org/10.1016/S0140-6736(14)60417-7
Ozaki K, Kadomoto R, Asato K, Tanimura S, Itoh N, Kohno M (2001) ERK pathway positively regulates the expression of Sprouty genes. Biochem Biophys Res Commun 285:1084–1088. https://doi.org/10.1006/bbrc.2001.5295
Purcell NH, Wilkins BJ, York A, Saba-El-Leil MK, Meloche S, Robbins J, Molkentin JD (2007) Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc Natl Acad Sci USA 104:14074–14079. https://doi.org/10.1073/pnas.0610906104
Rose BA, Force T, Wang Y (2010) Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev 90:1507–1546. https://doi.org/10.1152/physrev.00054.2009
Rossello X, Yellon DM (2017) The RISK pathway and beyond. Basic Res Cardiol 113:2. https://doi.org/10.1007/s00395-017-0662-x
Sanada S, Kitakaze M, Papst PJ, Hatanaka K, Asanuma H, Aki T, Shinozaki Y, Ogita H, Node K, Takashima S, Asakura M, Yamada J, Fukushima T, Ogai A, Kuzuya T, Mori H, Terada N, Yoshida K, Hori M (2001) Role of phasic dynamism of p38 mitogen-activated protein kinase activation in ischemic preconditioning of the canine heart. Circ Res 88:175–180. https://doi.org/10.1161/res.88.2.175
Schulz R, Belosjorow S, Gres P, Jansen J, Michel MC, Heusch G (2002) p38 MAP kinase is a mediator of ischemic preconditioning in pigs. Cardiovasc Res 55:690–700. https://doi.org/10.1016/S0008-6363(02)00319-X
Sharma B, Joshi S, Sassano A, Majchrzak B, Kaur S, Aggarwal P, Nabet B, Bulic M, Stein BL, McMahon B, Baker DP, Fukunaga R, Altman JK, Licht JD, Fish EN, Platanias LC (2012) Sprouty proteins are negative regulators of interferon (IFN) signaling and IFN-inducible biological responses. J Biol Chem 287:42352–42360. https://doi.org/10.1074/jbc.M112.400721
Sugden PH, Clerk A (1997) Regulation of the ERK subgroup of MAP kinase cascades through G protein-coupled receptors. Cell Signal 9:337–351. https://doi.org/10.1016/S0898-6568(96)00191-X
Sutherland C, Leighton IA, Cohen P (1993) Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 296:15–19. https://doi.org/10.1042/bj2960015
Thornton TM, Pedraza-Alva G, Deng B, Wood CD, Aronshtam A, Clements JL, Sabio G, Davis RJ, Matthews DE, Doble B, Rincon M (2008) Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science 320:667–670. https://doi.org/10.1126/science.1156037
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S (2008) MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456:980–984. https://doi.org/10.1038/nature07511
Yue TL, Wang C, Gu JL, Ma XL, Kumar S, Lee JC, Feuerstein GZ, Thomas H, Maleeff B, Ohlstein EH (2000) Inhibition of extracellular signal-regulated kinase enhances ischemia/reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res 86:692–699. https://doi.org/10.1161/res.86.6.692
Zhai P, Sciarretta S, Galeotti J, Volpe M, Sadoshima J (2011) Differential roles of GSK-3beta during myocardial ischemia and ischemia/reperfusion. Circ Res 109:502–511. https://doi.org/10.1161/CIRCRESAHA.111.249532
Acknowledgements
Open access funding provided by University of Oulu including Oulu University Hospital. We thank Marja Arbelius, Kirsi Salo and Sirpa Rutanen for excellent technical assistance. This work was supported by Academy of Finland Grant no. 256908 to J.U., 268505 to J.M., and Grants 131020 and 297094 to R.K., by the Finnish Foundation for Cardiovascular Research (to T.A., J.U., L.V., J.M., Z.S. and R.K.), by Finnish Cultural Foundation (T.A.), by Aarne Koskelo Foundation (T.A.), by Emil Aaltonen Foundation (J.U.), by Orion Research Foundation (J.U.), by Sigrid Juselius Foundation (R.K.) and by Jane and Aatos Erkko Foundation (R.K.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Alakoski, T., Ulvila, J., Yrjölä, R. et al. Inhibition of cardiomyocyte Sprouty1 protects from cardiac ischemia–reperfusion injury. Basic Res Cardiol 114, 7 (2019). https://doi.org/10.1007/s00395-018-0713-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-018-0713-y