
Abstract
Mutations in the phosphate-regulating endopeptidase homolog, X-linked, gene (PHEX), which encodes a zinc-dependent endopeptidase that is involved in bone mineralization and renal phosphate reabsorption, cause the most common form of hypophosphatemic rickets, X-linked hypophosphatemic rickets (XLH). The distribution of PHEX mutations is extensive, but few mutations have been identified in Chinese with XLH. We extracted genomic DNA and total RNA from leukocytes obtained from nine unrelated Chinese subjects (three males and six females, age range 11–36 years) who were living in Taiwan. The PHEX gene was amplified from DNA by PCR, and the amplicons were directly sequenced. Expression studies were performed by reverse-transcription PCR of leukocyte RNA. Serum levels of FGF23 were significantly greater in the patients than in normal subjects (mean 69.4 ± 18.8 vs. 27.2 ± 8.4 pg/mL, P < 0.005), and eight of the nine patients had elevated levels of FGF23. Germline mutations in the PHEX gene were identified in five of 9 patients, including novel c.1843 delA, donor splice site mutations c.663+2delT and c.1899+2T>A, and two previously reported missense mutations, p.C733Y and p.G579R. These data extend the spectrum of mutations in the PHEX gene in Han Chinese and confirm variability for XLH in Taiwan.
Similar content being viewed by others

Although acquired vitamin D deficiency is the principal cause of rickets worldwide, rickets (and osteomalacia) can also arise in patients with defects that perturb vitamin D metabolism or responsiveness or with conditions that cause hypophosphatemia. Phosphate homeostasis is principally controlled by changes in the tubular reabsorption of phosphate, which is regulated, directly or indirectly, by PTH, phosphatonins such as FGF23, and 1,25(OH)2D. Thus, hypophosphatemia and hypophosphatemic rickets most commonly occur in patients who have conditions that depress renal tubular phosphate transport. A variety of acquired and genetic disorders have been described in which decreased renal tubular phosphate reabsorption leads to the development of hypophosphatemic rickets. An acquired form of hypophosphatemia occurs in patients with fibrous dysplasia or mesenchymal tumors that overexpress FGF23, a phosphatonin that reduces expression of the NaPi2a and NaPi2c sodium–phosphate cotransporters in renal proximal tubular cells and impairs synthesis of 1,25(OH)2D [1, 2]. In addition, at least five inherited forms of hypophosphatemic rickets have now been characterized: X-linked hypophosphatemic rickets (XLH; MIM 307800, 307810) is caused by mutations in the PHEX gene located at Xp22.1 [3], autosomal dominant hypophosphatemic rickets (MIM 193100) is caused by mutations in the FGF23 gene [4], autosomal recessive hypophosphatemic rickets (MIM 600980) is caused by mutations in the DMP1 [5, 6] or ENPP1 [7, 8] gene, and autosomal recessive hypophosphatemic rickets with hypercalciuria (HHRH) is caused by mutations in SLC34A3 encoding the NaPi-IIc renal sodium–phosphate cotransporter [9–11]. XLH is the most prevalent form of inherited rickets in humans, with an incidence of 1 in 20,000 individuals [12, 13]. It is a dominant disorder characterized by hypophosphatemia secondary to renal phosphate wasting, abnormal regulation of renal metabolism of vitamin D, rickets and osteomalacia, growth failure, and resistance to vitamin D therapy. Despite marked hypophosphatemia, these patients seldom present with muscle weakness [13–15].
The PHEX gene encompasses 22 exons and encodes a 749-amino acid protein with significant structural homology to members of the type II integral membrane zinc-dependent endopeptidase family, with short N-terminal cytoplasmic domains, a single transmembrane domain, and a long extracellular domain [3]. Defective PHEX function is associated with increased serum concentrations of the phosphatonin FGF23, which likely accounts for the metabolic defects in hypophosphatemic rickets. More than 250 mutations in the PHEX gene have been identified to date in patients with XLH and are catalogued in a locus-specific database [16] (www.phexdb.mcgill.ca). The mutations, which are scattered throughout the gene, include deletions, splice junction and frameshift mutations, duplications, insertions, and missense and nonsense mutations and are consistent with loss of function of the PHEX protein. XLH due to unique mutations in PHEX has been reported in subjects of East Asian ethnicity including Japanese, Korean, and Chinese living in China and Taiwan [17–25]. Mutational founder effects for a variety of genetic disorders, as well as haplotype sharing in only a small chromosomal interval, have been found in East Asians that are not replicated in Caucasians, suggesting that specific mutations are ancient and occurred after the divergence of East Asians and Caucasians. In this study, we sought to identify PHEX gene mutations in Chinese patients with hypophosphatemic rickets who are living in Taiwan.
Patients and Methods
Patients
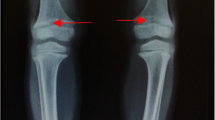
We studied nine unrelated Chinese subjects (three males and six females, age range 11–36 years) with clinical and biochemical manifestations of early-onset or congenital hypophosphatemic rickets, including hypophosphatemia, short stature, and lower extremity deformities (Table 1). There were three familial (cases 1, 2, and 5) that showed dominant transmission of rickets and six sporadic cases. At the time of these studies, all affected subjects except proband 1 and his sister and proband 2 and her brother were receiving treatment with an active form of vitamin D plus phosphate. This study was approved by the institutional review board of the Taipei Veterans General Hospital, and informed consent was obtained from each individual.
Serum Measurements
Serum concentrations of intact FGF23 were measured using a two-site enzyme-linked immunosorbent assay (FGF23 ELISA Kit; Kainos, Tokyo, Japan) according to the manufacturer’s recommendations. All samples were assayed at the same time in the same assay. Circulating levels of intact FGF-23 were measured in serum from nine patients, two siblings, and 11 age- and sex-matched normal individuals. Serum 1,25(OH)2D levels were measured with RIA (DiaSorin, Stillwater, MN). The TRP was calculated using the standard, formula and TmP/GFR was determined using the nomogram of Bijvoet [12, 26, 27].
PHEX Mutational Analysis
Genomic DNA was isolated from EDTA-preserved whole blood using the GFX Genomic Blood DNA purification kit (Amersham Biosciences, Piscataway, NJ). Twenty-two pairs of PHEX-specific primers [22] were used for PCR amplification of the coding exons and adjoining splice sites in a 25-μL reaction containing 200 ng of genomic DNA, 2.0 mM MgCl2, 0.2 mM of each dNTP, 0.15 M of each primer, 1× reaction buffer, and 1 unit of FastStart Taq DNA polymerase (Roche, Indianapolis, IN). The DNA sequences of both strands were determined by Taq polymerase cycle sequencing using fluorescence-labeled dideoxyterminators (BigDye Terminator V3.1 Cycle Sequencing Kits; Applied Biosystems, Foster City, CA) and resolved on an automated detection system (ABI 377-36 sequencer, Applied Biosystems). Mutations are reported according to the guidelines of the Human Genome Variation Society; based on the cDNA sequence, +1 is the A of the ATG start codon [28].
Novel mutations were defined by their absence from the Human Gene Mutation Database (http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html) and from mutations previously reported in the PHEX-specific database [16] (www.phexdb.mcgill.ca). The functional consequences of novel splice variants were predicted with the NNSPLICE 0.9 automated splice site analysis program on the web (http://www.fruitfly.org/seq_tools/splice.html) [29].
RT-PCR
Total RNA was extracted from peripheral whole blood using a QIAamp RNA Blood Mini Kit (Qiagen, Hilden, Germany). First-strand cDNA was generated with SUPERSCRIPT II reverse transcriptase (Invitrogen, Carlsbad, CA) using an oligo(dT) 12–18 primer and subsequently used as a template for PCR. For proband 2, the forward primer (ggtctcttaagtctccaagc) was located in exon 2 and the reverse primer (tttgttgtacatggcctcgc) was located in exon 8. PCR products of female subjects were cloned into pGEM-T vector (Promega, Madison, WI), and 10 clones were selected for sequence analysis (Applied Biosystems). For proband 3, the forward primer (atccgacgactgtcaatgcc) was located in exon 15 and the reverse primer (ttggacttgttctcgggcag) in exon 21. The products of RT-PCRs for cases 2 and 3 were electrophoresed on 3% agarose gel and stained with ethidium bromide. The identity of the amplicons was confirmed by nucleotide sequence analysis.
Statistical Analysis
All data are expressed as the mean ± SD, and comparison between groups was calculated using unpaired Student’s t-tests. Linear regression was used to compare phenotypic features and genotypes. P < 0.05 was considered significant. GraphPad InStat, version 3.06 (GraphPad, San Diego, CA), was used to perform statistical analyses.
Results
Clinical Features
The clinical, molecular, and biochemical data for all nine patients are shown in Table 1. Patients 4 and 5 had experienced multiple fractures of the lower extremities. The mean serum FGF23 level was significantly higher in patients than in normal subjects (69.4 ± 18.8 vs. 27.2 ± 8.4 pg/mL, P < 0.005) and frankly elevated in eight of nine patients (Table 1). At the time of sampling, probands 1 and 2 were not taking either calcitriol or phosphate; other subjects either were taking low doses of calcitriol and phosphate or were poorly compliant.
Mutations of the PHEX Gene
DNA analysis of the entire coding region together with the associated splice sites and 5′ and 3′ untranslated regions of the PHEX gene disclosed mutations in five of the 9 patients we studied (Table 1, Fig. 1). Patient 1 was hemizygous for a single nucleotide deletion (c.1843 del A) in exon 18 of the PHEX gene, resulting in a frameshift and early translational termination that is predicted to produce a protein that is truncated at amino acid residue 617. Patient 2 was heterozygous for a donor site mutation (c.663 + 2delT) in intron 5. RT-PCR of RNA from patient 2 produced two amplicons, one that corresponded to the expected size of 771 bp and a shorter amplicon of 694 bp (Fig. 1b). Analysis of this mutation by NNSPLICE 0.9 [29] predicted aberrant splicing of the PHEX mRNA, which was confirmed by RT-PCR of leukocyte RNA and nucleotide sequence analysis, which confirmed the usage of an alternative 5′-splice site in exon 5 that was located 77 nucleotides upstream of the normal donor splice site (Fig. 1). Patient 3 was hemizygous for a donor site transversion (c.1899+2T>A) in intron 18 that also was predicted to result in aberrant splicing of the PHEX mRNA. RT-PCR of leukocyte RNA from this patient revealed a PHEX cDNA that utilized an alternative 5′-splice site that is 131 nucleotides upstream of the normal donor site and results in skipping of exon 18 (Fig. 1c). The amplicon of RT-PCR corresponding to wild-type cDNA was 521 bp and the mutant transcript was 390 bp (Fig. 1c). These changes are predicted to result in a frameshift and a premature termination codon. None of these sequence changes was present in genomic DNA from 50 unrelated subjects.

Molecular analysis of PHEX. a Proband 1 had a c.1843 delA mutation in his copy of the PHEX gene. b Proband 2 was heterozygous for c.663+2delT, resulting in alternative splicing of mRNA from this allele. The amplicons generated by RT-PCR of RNA from a normal subject and the patient are shown and indicate that the patient had a smaller band (694 bp) than expected (771 bp) for the wild-type allele. The RT-PCR product of this patient was sequenced, and the 5′-splice site of intron 5 was shifted 77 nucleotides upstream in exon 5. c Patient 3 was hemizygous for the c.1899+2T>A mutation that resulted in an abnormally spliced mRNA in which the 5′-donor splice site of intron 18 was shifted 131 nucleotides upstream of the native donor splice site, resulting in skipping of exon 18. The amplicon corresponding to wild-type cDNA is 521 bp, and the corresponding mutant amplicon is 390 bp. d Patient 4 had a C733Y missense mutation (TGT → TAT) in codon 733 of exon 22. e Patient 5 had a G579R missense mutation (GGA → AGA) in codon 579 of exon 17
Patients 4 (C733Y) and 5 (G579R) were heterozygous for missense mutations that have been previously described [30, 31].
Patients 1, 2, and 5 have familial hypophosphatemic rickets. The affected brother and mother of proband 2 had the same genotype, and all of them had severe genu varum and short stature. Proband 2 had severe genu varum and had undergone osteotomy of the lower extremities four times. All of the clinically affected relatives of proband 5 carried the same mutation and presented with genu varum and short stature. Regrettably, neither of the two affected family members of proband 1 agreed to participate in this study.
There was no relationship between height SD and presence or absence of a PHEX mutation, nor were there any relationships between type of mutation and height SD, serum concentration of FGF23, and serum phosphate levels. Moreover, we found no evidence for a relationship between serum concentrations of FGF23 and serum levels of phosphate or calcitriol.
Discussion
We analyzed the PHEX gene in nine unrelated Chinese subjects with early-onset hypophosphatemic rickets, including three patients with familial rickets and six patients with sporadic rickets. Our mutation detection rates in the PHEX gene were 100% in familial cases and 33% in sporadic cases, which are similar to the results of most previous studies in which the mutation detection rates ranged 51–86% in familial cases and 22–57% in sporadic cases [18, 30–34]. One notable exception is a single study from Finland, in which the mutation detection rate was 100% for familial cases and 93% for sporadic cases [35]. The lower mutation detection rate in apparently sporadic cases might reflect nongenetic causes of renal phosphaturia, autosomal recessive forms of hypophosphatemic rickets due to mutations in the DMP1 [5, 6] or ENPP1 [7, 8] gene, or the presence of de novo mutations in the FGF23 gene [4].
Our work raises several points that are worth noting. First, all of the PHEX mutations that we identified in this study were different from the mutations that have been previously described in Chinese patients with XLH [19] (Fig. 2), indicating that there is significant variability in PHEX mutations in this population. Second, the mutations in the coding exons of PHEX were found in the 3′ end of the gene, which is consistent with the results of previous studies [36, 37] and mutations entered in PHEXdb [16], which suggests that this region might encode for a critical region of the PHEX protein. Third, the two donor splice site mutations both resulted in an aberrant 5′-splice site. As is typical of mutations at donor or acceptor sites, the two mutations that we identified utilized upstream cryptic splice sites that in one case resulted in skipping of the upstream exon. Mutations affecting splice sites are common in human genetic disease and account for 10–15% of gene mutations. The majority of these are single-point mutations affecting the conserved bases at the donor or acceptor splice sites. A study of more than 100 splice-site mutations [38, 39] showed that point mutations affecting the 5′ donor splice site were more common than those at the 3′ acceptor site (62% vs. 26%). At the 5′ donor splice site, mutations affecting the G residue at position +1 are the most common, followed by mutations at position +5. Mutations at these two positions are thought to significantly reduce the pairing of the donor splice site with the complementary site in the small nuclear ribonucleoprotein particle U1snRNP, which is one of the first steps in the complex process of mRNA splicing [40, 41].

The distribution of PHEX gene mutations among Chinese with XLH is shown. Mutations in exon 18, in which the zinc-binding domain is located, are the most common and account for 30% (4/13) of PHEX mutations in Chinese subjects
Lastly, we found elevated serum levels of intact FGF23 in eight of nine patients with hypophosphatemic rickets. Our results confirm the association of elevated circulating FGF23 and hypophosphatemia in subjects with XLH and other disorders characterized by hypophosphatemia [2, 42–44]. Recent studies show that most subjects with XLH demonstrate increases in serum FGF23 in response to treatment with calcitriol and phosphate, with significant and nonsignificant positive associations with daily phosphate and calcitriol doses, respectively [45]. However, the relatively low doses of calcitriol and phosphorus used by the mostly adult subjects in our study, as well as generally poor compliance in our population, did not permit us to identify similar associations. Moreover, the cross-sectional design of our study makes it very difficult to determine if an association or a lack of such is caused by differences in treatment or by primary differences in the phenotype.
The sodium–phosphate cotransporters NaPi-IIc and NaPi-IIa are coexpressed in the renal brush border membrane of the proximal tubule, where most filtered phosphate is reabsorbed. NaPi-IIa and NaPi-IIc are both regulated by PTH, FGF23, and dietary phosphate; PTH and FGF23, in response to elevated levels of dietary and serum phosphorus, decrease the abundance of cotransporters at the cell membrane and thereby reduce the tubular reabsorption of phosphorus from the glomerular filtrate [9, 46, 47]. Serum levels of FGF23 are inappropriately normal or elevated in XLH and other forms of hypophosphatemic rickets [1, 2] except ADHR [11, 48].
A limitation of our study is the small sample size, which may have restricted our ability to identify potential genotype–phenotype relationships between XLH and any of the mutations that we identified. Although Holm et al. [31] identified a trend between truncating mutations and more severe skeletal disease in a familial group (P = 0.072), at present no consensus has been reached regarding the correlation between the disease phenotypes and PHEX mutation locations, including those in this study [16, 18, 20, 22, 33, 37, 49, 50].
In conclusion, we have identified five mutations in the PHEX gene in Han Chinese patients with XLH in Taiwan, including three novel mutations. Our results, taken in the context of prior studies of PHEX gene analyses in East Asians, confirm the notion that variable PHEX mutations account for XLH in this population and extend the spectrum of PHEX gene defects that cause XLH.
References
Fukumoto S, Yamashita T (2007) FGF23 is a hormone-regulating phosphate metabolism—unique biological characteristics of FGF23. Bone 40:1190–1195
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 348:1656–1663
Consortium HYP (1995) A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet 11:130–136
(2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM (2006) DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet 38:1248–1250
Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, Parvari R (2010) Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet 86:273–278
Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM (2010) Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet 86:267–272
Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, Frappier D, Burkett K, Carpenter TO, Anderson D, Garabedian M, Sermet I, Fujiwara TM, Morgan K, Tenenhouse HS, Juppner H (2006) SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet 78:179–192
Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, Econs MJ (2006) Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab 91:4022–4027
Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P, Schnabel D, Hochberg Z, Strom TM (2006) Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium–phosphate cotransporter gene SLC34A3. Am J Hum Genet 78:193–201
Strom TM, Juppner H (2008) PHEX, FGF23, DMP1 and beyond. Curr Opin Nephrol Hypertens 17:357–362
Tenenhouse HS, Econs MJ (2001) Mendelian hypophosphatemias. In: Scriver CR, Beaudet AL, Sly WS, Vale D (eds) The metabolic and molecular basis of inherited disease. McGraw Hill, New York, pp 5039–5068
Makras P, Hamdy NA, Kant SG, Papapoulos SE (2008) Normal growth and muscle dysfunction in X-linked hypophosphatemic rickets associated with a novel mutation in the PHEX gene. J Clin Endocrinol Metab 93:1386–1389
Glorieux F, Karsenty G, Thakker RV (1997) Metabolic bone disease in children. In: Avioli LV, Krane SM (eds) Metabolic bone diseases and clinically related disorders. Academic Press, New York, pp 759–783
Sabbagh Y, Jones AO, Tenenhouse HS (2000) PHEXdb, a locus-specific database for mutations causing X-linked hypophosphatemia. Hum Mutat 16:1–6
Saito T, Nishii Y, Yasuda T, Ito N, Suzuki H, Igarashi T, Fukumoto S, Fujita T (2009) Familial hypophosphatemic rickets caused by a large deletion in PHEX gene. Eur J Endocrinol 161:647–651
Cho HY, Lee BH, Kang JH, Ha IS, Cheong HI, Choi Y (2005) A clinical and molecular genetic study of hypophosphatemic rickets in children. Pediatr Res 58:329–333
Xia W, Meng X, Jiang Y, Li M, Xing X, Pang L, Wang O, Pei Y, Yu LY, Sun Y, Hu Y, Zhou X (2007) Three novel mutations of the PHEX gene in three Chinese families with X-linked dominant hypophosphatemic rickets. Calcif Tissue Int 81:415–420
Song HR, Park JW, Cho DY, Yang JH, Yoon HR, Jung SC (2007) PHEX gene mutations and genotype–phenotype analysis of Korean patients with hypophosphatemic rickets. J Korean Med Sci 22:981–986
Lo FS, Kuo MT, Wang CJ, Chang CH, Lee ZL, Van YH (2006) Two novel PHEX mutations in Taiwanese patients with X-linked hypophosphatemic rickets. Nephron Physiol 103:157–163
Chou YY, Chao SC, Tsai SC, Lin SJ (2005) Novel PHEX gene mutations in two Taiwanese patients with hypophosphatemic rickets. J Formos Med Assoc 104:198–202
Goji K, Ozaki K, Sadewa AH, Nishio H, Matsuo M (2006) Somatic and germline mosaicism for a mutation of the PHEX gene can lead to genetic transmission of X-linked hypophosphatemic rickets that mimics an autosomal dominant trait. J Clin Endocrinol Metab 91:365–370
Sato K, Tajima T, Nakae J, Adachi M, Asakura Y, Tachibana K, Suwa S, Katsumata N, Tanaka T, Hayashi Y, Abe S, Murashita M, Okuhara K, Shinohara N, Fujieda K (2000) Three novel PHEX gene mutations in Japanese patients with X-linked hypophosphatemic rickets. Pediatr Res 48:536–540
Su P-H, Chen Y-J, Chen S-J, Yu J-S, Yieh D-M (2010) Novel PHEX gene mutation in one Taiwanese woman with hypophosphatemic rickets. Korean J Genet 29:37–42
Alon U (1993) Clinical assessment of plasma phosphate and renal tubular threshold for phosphate. In: Alon U, Chan JCM (eds) Phosphate in pediatric health and disease. CRC Press, Ann Arbor, pp 103–114
Brodehl J, Krause A, Hoyer PF (1988) Assessment of maximal tubular phosphate reabsorption: comparison of direct measurement with the nomogram of Bijvoet. Pediatr Nephrol 2:183–189
den Dunnen JT, Antonarakis SE (2000) Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 15:7–12
Reese MG, Eeckman FH, Kulp D, Haussler D (1997) Improved splice site detection in Genie. J Comput Biol 4:311–323
Dixon PH, Christie PT, Wooding C, Trump D, Grieff M, Holm I, Gertner JM, Schmidtke J, Shah B, Shaw N, Smith C, Tau C, Schlessinger D, Whyte MP, Thakker RV (1998) Mutational analysis of PHEX gene in X-linked hypophosphatemia. J Clin Endocrinol Metab 83:3615–3623
Holm IA, Nelson AE, Robinson BG, Mason RS, Marsh DJ, Cowell CT, Carpenter TO (2001) Mutational analysis and genotype–phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab 86:3889–3899
Rowe PS, Oudet CL, Francis F, Sinding C, Pannetier S, Econs MJ, Strom TM, Meitinger T, Garabedian M, David A, Macher MA, Questiaux E, Popowska E, Pronicka E, Read AP, Mokrzycki A, Glorieux FH, Drezner MK, Hanauer A, Lehrach H, Goulding JN, O’Riordan JL (1997) Distribution of mutations in the PEX gene in families with X-linked hypophosphataemic rickets (HYP). Hum Mol Genet 6:539–549
Gaucher C, Walrant-Debray O, Nguyen TM, Esterle L, Garabedian M, Jehan F (2009) PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet 125:401–411
Francis F, Strom TM, Hennig S, Boddrich A, Lorenz B, Brandau O, Mohnike KL, Cagnoli M, Steffens C, Klages S, Borzym K, Pohl T, Oudet C, Econs MJ, Rowe PS, Reinhardt R, Meitinger T, Lehrach H (1997) Genomic organization of the human PEX gene mutated in X-linked dominant hypophosphatemic rickets. Genome Res 7:573–585
Tyynismaa H, Kaitila I, Nanto-Salonen K, Ala-Houhala M, Alitalo T (2000) Identification of fifteen novel PHEX gene mutations in Finnish patients with hypophosphatemic rickets. Hum Mutat 15:383–384
Filisetti D, Ostermann G, von Bredow M, Strom T, Filler G, Ehrich J, Pannetier S, Garnier JM, Rowe P, Francis F, Julienne A, Hanauer A, Econs MJ, Oudet C (1999) Non-random distribution of mutations in the PHEX gene, and under-detected missense mutations at non-conserved residues. Eur J Hum Genet 7:615–619
Ichikawa S, Traxler EA, Estwick SA, Curry LR, Johnson ML, Sorenson AH, Imel EA, Econs MJ (2008) Mutational survey of the PHEX gene in patients with X-linked hypophosphatemic rickets. Bone 43:663–666
Krawczak M, Reiss J, Cooper DN (1992) The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 90:41–54
Cooper DN, Krawczak M, Antonarakis SE (1995) The nature and mechanisms of human gene mutation. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 259–291
Kramer A (1996) The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem 65:367–409
Wittop Koning TH, Schumperli D (1994) RNAs and ribonucleoproteins in recognition and catalysis. Eur J Biochem 219:25–42
Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S (2002) Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 87:4957–4960
Weber TJ, Liu S, Indridason OS, Quarles LD (2003) Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res 18:1227–1234
Ito N, Fukumoto S, Takeuchi Y, Yasuda T, Hasegawa Y, Takemoto F, Tajima T, Dobashi K, Yamazaki Y, Yamashita T, Fujita T (2005) Comparison of two assays for fibroblast growth factor (FGF)-23. J Bone Miner Metab 23:435–440
Imel EA, DiMeglio LA, Hui SL, Carpenter TO, Econs MJ (2010) Treatment of X-linked hypophosphatemia with calcitriol and phosphate increases circulating fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab 95:1846–1850
Ferrari SL, Bonjour JP, Rizzoli R (2005) Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab 90:1519–1524
Antoniucci DM, Yamashita T, Portale AA (2006) Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab 91:3144–3149
Tencza AL, Ichikawa S, Dang A, Kenagy D, McCarthy E, Econs MJ, Levine MA (2009) Hypophosphatemic rickets with hypercalciuria due to mutation in SLC34A3/type IIc sodium–phosphate cotransporter: presentation as hypercalciuria and nephrolithiasis. J Clin Endocrinol Metab 94:4433–4438
Clausmeyer S, Hesse V, Clemens PC, Engelbach M, Kreuzer M, Becker-Rose P, Spital H, Schulze E, Raue F (2009) Mutational analysis of the PHEX gene: novel point mutations and detection of large deletions by MLPA in patients with X-linked hypophosphatemic rickets. Calcif Tissue Int 85:211–220
Ellison J, Tebben P (2009) Novel human pathological mutations. Gene symbol: PHEX. Disease: rickets, hypophosphataemic. Hum Genet 125:339
Acknowledgements
This study was supported by grants from Medical Research Council (VGH-97-C1-002) and National Science Council (NSC-96-2314-B-075-018-MY3), Taiwan, ROC, and generous gifts from the Friedman Family Foundation and Sadikoglu family.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors have stated that they have no conflict of interest.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Jap, TS., Chiu, CY., Niu, DM. et al. Three Novel Mutations in the PHEX Gene in Chinese Subjects with Hypophosphatemic Rickets Extends Genotypic Variability. Calcif Tissue Int 88, 370–377 (2011). https://doi.org/10.1007/s00223-011-9465-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-011-9465-5