
Abstract
Despite significant advances in early detection and treatment, breast cancer remains a major cause of morbidity and mortality. Perfluorooctanoic acid (PFOA) is a suspected endocrine disruptor and a common environmental pollutant associated with various diseases including cancer. However, the effects of PFOA and its mechanisms of action on hormone-responsive cells remain unclear. Here, we explored the potential tumorigenic activity of PFOA (100 nM–1 mM) in human breast epithelial cells (MCF-10A). MCF-10A cells exposed to 50 and 100 µM PFOA demonstrated a higher growth rate compared to controls. The compound promoted MCF-10A proliferation by accelerating G0/G1 to S phase transition of the cell cycle. PFOA increased cyclin D1 and CDK4/6 levels, concomitant with a decrease in p27. In contrast to previous studies of perfluorooctane sulfate (PFOS), the estrogen receptor antagonist ICI 182,780 had no effect on PFOA-induced cell proliferation, whereas the PPARα antagonist GW 6471 was able to prevent the MCF-10A proliferation, indicating that the underlying mechanisms involve PPARα-dependent pathways. Interestingly, we also showed that PFOA is able to stimulate cell migration and invasion, demonstrating its potential to induce neoplastic transformation of human breast epithelial cells. These results suggest that more attention should be paid to the roles of PFOA in the development and progression of breast cancer.
Similar content being viewed by others
Introduction
Breast cancer is one of the most common malignancies that women in Western countries may develop in their lifetime (Gullick et al. 1998). During the last decades, the breast cancer incidence has globally increased and accounts for over 20% of the total cancer cases and about 14% of cancer deaths among females (Jemal et al. 2011). Despite extensive research efforts to understand and treat breast cancer, risk factors and cellular processes that can lead to the onset of mammary carcinogenesis have yet to be elucidated in detail. About 5–10% of the cases are due to inheritance of risk genes such as BRCA1 and BRCA2 (Campeau et al. 2008). Several common persistent organic pollutants are endocrine disrupters and proposed to play an important role in cancer etiology. These compounds have been linked to effects relevant for the development of breast cancer, including tumor promoting, and immunosuppressive activities (Bonefeld-Jorgensen et al. 2011; Kortenkamp 2006). Among them, both estrogenic and non-estrogenic endocrine disruptors are considered to play critical roles in human breast carcinogenesis (Brody et al. 2007). Hence, studies of hormonally active compounds contaminating the environment, food and water are a priority.
In recent years, human and wildlife monitoring studies have ubiquitously detected manmade perfluoroalkyl acids (PFAAs) throughout the world. These chemicals are frequently used in industrial and consumer products, because of their stain-resistant and water-repellant characteristics. The two most widely known PFAAs are perfluorooctanoic acid (PFOA), and perfluorooctane sulfate (PFOS) (Lau et al. 2007). PFOA has been used in products such as non-stick cookware, cosmetics, upholstery, and is known to bio-accumulate in food chains (Bartell et al. 2010). In addition, PFOA does not break down in the environment and can be found in drinking water at concentrations ranging from picograms to micrograms per liter (Rayne and Forest 2009; Shin et al. 2011). PFOA has also been detected in blood of most people examined in a number of industrialized countries. The exposure levels vary among populations and over time (Bartell et al. 2010; Herrick et al. 2017). In blood, PFOA has been reported to reach values between 7 and 9370 ng/mL, whereas the median level for the general US population is considered to be approximately 5 ng/mL (Emmett et al. 2006; Steenland et al. 2010). The compound is not metabolized in the body, and the human half-life is estimated between 4 and 5 years (Emmett et al. 2006; Lau et al. 2007). According with this, a study using post-mortem samples found concentrations around 3.8 and 1.4 ng/g in liver and adipose tissue, respectively (Maestri et al. 2006).
PFOA exposure may lead to a variety of adverse effects, including hepatotoxicity, immunotoxicity, and developmental toxicity (Lau et al. 2007; Yang et al. 2000). Moreover, toxicologic data show that dietary intake of PFOA induces tumors of various tissues in rodents (Biegel et al. 2001). Animal studies have also indicated that PFOA exposure affects mammary gland development and increases mammary fibroadenomas (White et al. 2007). Recent epidemiological studies support a possible association between both PFOS and PFOA and tumorigenesis, including liver, pancreatic, and testicular cancer (Barry et al. 2013; Lau et al. 2007; Vieira et al. 2013), as well as breast cancer (Bonefeld-Jorgensen et al. 2011; Wielsoe et al. 2017). PFOS and PFOA are suspected to be endocrine disruptors with estrogenic activity, but the potential for these environmental contaminants to induce estrogenic disruption in mammals is not fully known (Jensen and Leffers 2008; Sonthithai et al. 2016).
Mechanisms by which PFAAs may affect breast tissue can be studied in animal and cell models. MCF-10A is a subline of spontaneously immortalized human breast epithelial cells—derived from human fibrocystic mammary tissue—with characteristics of normal breast epithelium (Soule et al. 1990). These features make the MCF-10A a valuable in vitro model for studies of normal breast cell function and determine the potential of environmental contaminants to induce tumor transformation. We have recently demonstrated that PFOS is capable of transforming MCF-10A to a malignant profile by altering important cell cycle proteins as well as inducing cell migration and invasion (Pierozan and Karlsson 2018).
In this study, we evaluate the potential tumorigenic activity of PFOA in MCF-10A cells. Cell viability, cell counting, and cell cycle analysis were applied to investigate the effects of PFOA exposure on cell proliferation. To better understand the response of MCF-10A cells to PFOA exposure, we studied the regulatory cell cycle proteins, cell migration/invasion and estrogen receptor (ER), pregnane X receptor (PXR) and peroxisome proliferator-activated receptor alpha (PPARα) activation. The findings of this study will help clarify the mechanisms underlying possible effects of PFOA on breast cancer development.
Materials and methods
Chemicals
Dimethyl sulfoxide (DMSO), paraformaldehyde, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), Triton X-100, propidium iodide (PI), DNAse-free RNAse A, perfluorooctanoic acid, cholera toxin (CT), insulin, 3-(4,5-dimethyl-2-yl)2,5-diphenyl-2H-tetrazolium bromide (MTT), epidermal growth factor (EGF), hydrocortisone and sulforaphane were obtained from Sigma-Aldrich (St Louis, MO, USA). Horse serum, penicillin–streptomycin (P/S), Dulbecco’s Phosphate-Buffered Saline (PBS), Dulbecco’s Modified Eagle’s Medium (DMEM), trypsin solution (0.05%) were obtained from Gibco (Invitrogen, Paisley, UK). p53 monoclonal (DO-7), CDK6 monoclonal (75B9), CDK4 monoclonal (DCS-31) and p21 monoclonal (R.229.6) antibodies were obtained from ThermoFisher Scientific (Rockford, IL, USA). P27 Kip1 (D69C12) and cyclin D1 (92G2) antibodies were obtained from Cell Signaling (Danvers, MA, USA). The secondary antibodies Alexa-Fluor 555 goat anti-mouse IgG or 488 goat anti-rabbit IgG, and the blocking agent (normal goat serum) were obtained from Molecular Probes, Invitrogen. Matrigel Basement Membrane Matrix was obtained from Corning (New York, NY, USA). ER α (sc-8002) and ERβ (sc-8974) monoclonal antibodies were obtained from Santa Cruz Biotechnology (Bergheimer, HD, DE). ICI 182,780, GW 6471 was obtained from Tocris Bioscience (Avonmouth, Bristol, UK).
Cell culture
MCF-10A cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were maintained as a monolayer in 10-cm2 tissue culture plastic flasks containing 10 mL of growth medium, trypsinized (0.05%) and split 1:5 every 3 days. Complete growth medium consisted of Dulbecco’s Modifies Eagle Medium with F-12 (DMEM/F-12; GIBCO, Invitrogen, Paisley, UK) supplemented with horse serum (5%), EGF (20 ng/mL), hydrocortisone (0.5 mg/mL), CT (100 ng/mL), insulin (10 mg/mL), and 5 mL P/S. Cell cultures were maintained at 37 °C and 5% CO2 in a humidified incubator.
Exposure of MCF-10A cells to PFOA
MCF-10A cells were trypsinized and resuspended in growth medium, plated in 96-well tissue culture plates (2 × 104 cells/well), and allowed to attach for 24 h in a 5% CO2 humidified incubator at 37 °C. After 24 h, the cells were treated with different PFOA concentrations (0–1 mM) dissolved in water and assay medium (growth medium without horse serum and EGF). The cells were incubated for 24, 48 and 72 h. All experiments were repeated three times.
MTT cell viability assay
MCF-10A cells were treated with different concentrations of PFOA (0–1 mM) for 24, 48 and 72 h, using eight wells for each treatment and three independent experiments. Cell viability was measured by the MTT assay. In brief, 0.5 mg MTT was added to each well of the 96-well plates (containing 100 µL medium and cells) 1 h before the end of incubation with PFOA. The supernatant was then removed and 100 µL DMSO was added to each well, followed by incubation and shaking for 10 min. The formazan product generated during the incubation was solubilized in DMSO and measured at 490 and 630 nm using a Polarstar Optima microplate reader (Bmg Labtech, Offenburg, Germany).
To investigate the participation of receptors on the PFOA effects, the cells were preincubated with the ER receptor blocker ICI 182,780 (100 nM) (Wrobel and Gregoraszczuk 2014), the PXR antagonist sulforaphane (1 µM) (Zhou et al. 2008) and the PPARα blocker GW6471 (1 µM) (Hah et al. 2014) for 30 min, before treatment with PFOA 100 µM for 72 h, and the MTT assay.
Cell counting by DAPI staining
MCF-10A cells were treated with different concentrations of PFOA (0–1 mM) for 24, 48 and 72 h, with three replicates of each treatment. After the treatment, cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton X-100 in PBS for 5 min at room temperature. Cells were stained with DAPI (0.25 mg/mL) for 10 min at room temperature followed by two washes with PBS. Cells were viewed in an ImageXpress Micro XLS Widefield High-Content Analysis System (Molecular Devices, Sunnyvale CA, USA), and images analyzed with the SoftMax Pro Software after digital acquisition (Molecular Devices, Sunnyvale CA, USA).
Analysis of cell cycle phases and proteins involved in cell cycle regulation
Cells were treated for 24, 48 and 72 h with 100 µM PFOA and processed for PI staining and flow cytometry as described previously (Pozarowski and Darzynkiewicz 2004). Forward and light scatter data were collected in a linear mode. Fluorescence data for 10,000 cells per sample were collected in the FL3 channel on a linear scale. Side- and forward-light scatter parameters were used to identify the cell events and doublets were excluded using gating. Samples were analyzed using a Cytoflex flow cytometer (Beckman Coulter Ltd., Brea, CA, USA). Cells in different cell cycle phases were presented as a percentage of the total number of cells counted.
To evaluate the effects on proteins involved in cell cycle regulation, cells were incubated with anti-cyclin D, anti-CDK4/6, anti-p21/27/53 antibodies and analyzed by flow cytometry using a Cytoflex flow cytometer (Beckman Coulter Ltd., Brea, CA, USA) as described previously (Pierozan and Karlsson 2018). Side and forward scatter of aggregates or lysed cells were determined using log scale SSC/FSC plots with thresholds. Voltage settings for the SSC, FSC and the fluorescent filters were kept constant for all experiments described. The results are expressed as mean fluorescence intensity compared with the controls (% of controls) for 10,000 cells per sample.
Immunocytochemistry
MCF-10A cells were treated with 100 µM PFOA 72 h and immunocytochemistry was performed as previously described (Pierozan et al. 2016). Negative control reactions were performed by omitting the primary antibody with no observed fluorescence. Cells were examined in an Olympus inverted microscope BX53 (Olympus, Tokyo, Japan), and images were collected with a 20 × objective using constant intensity settings and exposure time for all samples. The intensity of the cell fluorescence was measured using the image J software (ImageJ2), and the fluorescence intensity was estimated as the difference between the measured fluorescence of the cells and the background.
Migration and invasion assay
Transwell migration and invasion assays were conducted in 96-well plates with 8 µm pore-size membrane inserts (Corning, New York, NY, USA). Cells were treated with 100 µM PFOA for 72 h. After that, 5 × 105 cells were resuspended in 50 µL of assay medium and seeded in the upper chamber of transwells with (invasion assay) or without (migration assay) Matrigel Matrix (200 µg/mL). The lower chamber contained 100 µL growth medium. Cells were incubated for 24 h at 37 °C in a humidified atmosphere with 5% CO2. At the end of incubation, noninvasive cells in the upper chamber were removed and invasive cells in the bottom were fixed with 4% formaldehyde and stained with DAPI and counted as described above using an ImageXpress Micro XLS Widefield High-Content Analysis System (Molecular Devices, Sunnyvale, CA, USA), and the SoftMax Pro Software (Molecular Devices, Sunnyvale, CA, USA).
Western blot analysis
For evaluation of ER protein levels, cells were exposed to 100 µM PFOA or 10 nM 17β-estradiol (E2; positive control) for 72 h. Cells were then washed with ice-cold PBS and lysed with Laemmli lysis buffer. The protein concentration in cell lysates was determined using bicinchoninic acid protein assay (Smith et al. 1985). Equal amounts of protein (30 µg) were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on 10% gel and transferred (Mini Trans-Blot Electrophoretic Transfer Cell; Bio-Rad, Hercules, CA, USA) to polyvinylidene difluoride membranes for 1 h at 100 V in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol and 0.1% SDS). The blot was then washed for 20 min in Tris-buffered saline (TBS; 500 mM NaCl, 20 mM Trizma, pH 7.5), followed by 2 h incubation in blocking solution (TBS plus 5% defatted dry milk). After the incubation, the blot was washed twice for 5 min with blocking solution containing 0.05% Tween-20 (T-TBS) and incubated overnight at 4 °C in blocking solution containing monoclonal antibodies diluted 1:1000. The blot was then washed twice for 5 min with T-TBS and incubated for 2 h in a solution containing peroxidase-conjugated rabbit anti-mouse IgG diluted 1:2000 or peroxidase-conjugated anti-rabbit IgG diluted 1:2000. The blot was washed twice with T-TBS for 5 min and twice with TBS for 5 min. The blot was developed with the chemiluminescence ECL kit (Little Chalfont, UK).
Statistical analysis
The results were presented as means ± standard deviation (SD) for each experimental group of at least three individual samples. Differences between the groups were analyzed by one-way analysis of variance (ANOVA) followed by Tukey–Kramer multiple test, or by Student’s t test when comparing only two groups, using Graphpad Prism 7 software.
Results
PFOA-induced cell death and proliferation are dependent on the time and concentration
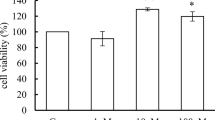
We first studied the effects of PFOA exposure on MCF-10A viability. Cells were incubated with 0–1 mM PFOA for 24, 48 and 72 h, and the cell viability determined by the MTT assay. The results showed that exposure to PFOA at 50 and 100 µM for 72 h increased the MTT production (Fig. 1c). In contrast, exposure to concentrations equals to 250 µM or higher decreased cell viability at all time points (Fig. 1a–c). To confirm these results, we determined the number of cells using DAPI staining. PFOA increased the number of cells at the concentrations of 50 and 100 µM at 48–72 h exposure (Fig. 1e, f), while the compound caused a decrease in the number of cells in the concentrations from 250 µM and higher at all time points (Fig. 1d–f).

Effects of PFOA on the viability of MCF-10A cells. The cells were exposed to 0–1 mM PFOA for 24, 48 and 72 h. The viability was determined by MTT assay (a–c) and DAPI staining (d–f). Values represent mean ± SD from three independent experiments. Statistically significant differences from control are indicated as follows: ***p < 0.001 and *p < 0.05 (one-way ANOVA followed by the Tukey–Kramer test)
PFOA alters the cell cycle in MCF-10A cells
Since we found an increase in cell proliferation of cells treated with 50–100 µM PFOA, we determined the effects of PFOA exposure on cell cycle distribution using PI staining and flow cytometry analysis. Table 1 shows the mean percentage of gated cells for each DNA content fraction (G0/G1, S, G2/M) following 24, 48 and 72 h exposure to 100 µM PFOA. The results demonstrated that PFOA exposure significantly reduced the percentage of cells in G0/G1 phase and increased the percentage of cells in S phase at all time points.
The levels of proteins involved in cell cycle regulation are altered by PFOA
To investigate the mechanisms involved in PFOA-induced cell proliferation and the alteration of the cell cycle in MCF-10A cells, the levels of the cyclin-dependent kinases (CDKs) CDK4, CDK6, cyclin D1 and their respective inhibitors (p27, p21 and p53) were analyzed by immunocytochemistry and flow cytometry. The fluorescence microscopy images revealed a reduced p27 level (Fig. 2a, b) and increased CDK6 (Fig. 2a, c), CDK4 and cyclin D levels (Fig. 2d–m), with no alteration on p21 and p53 levels (Fig. 2g–o). Confirming these results, flow cytometry analysis showed a decrease in the mean fluorescence intensity in p27-staining (Fig. 2j), and an increase in the fluorescence intensity in CDK6, CDK4 and cyclin D staining (Figure 2k–m) in PFOA-treated cells compared to the control group.

Effects of PFOA on the levels of proteins involved in cell cycle regulation. The cells were exposed to 100 µM PFOA for 72 h before immunocytochemistry and flow cytometry was performed. Representative images of PFOA-treated cells immunostained with p27 and CDK6 (a), cyclin D1 and CDK4 (b) and p21 and p53 (c). Mean fluorescence intensity was analyzed with immunocytochemistry (b–i) and flow cytometry (j–o) as described in “Material and methods” section. Values represent mean ± SD from three independent experiments. Scale bar = 50 µm. Statistically significant differences from control are indicated as follows: ***p < 0.001; **p < 0.01 and *p < 0.05 (Student’s t test)
PFOA exposure stimulates MCF-10A migration and invasion
To examine the involvement of PFOA on cell aggression, we performed a transwell migration and matrigel invasion assays. PFOA treatment at 100 µM significantly promoted cell migration and invasion of MCF-10A cells (Fig. 3a, b), suggesting that PFOA can induce MCF-10A transformation.

Effects of PFOA on MCF-10A cell migration and invasion capacity. Effects of PFOA on MCF-10A cell migration (a) and cell invasion (b) by a transwell assay. Migrated or invaded cells in the bottom were fixed with 4% formaldehyde and stained with DAPI and counted as described in the “Material and methods” section. Values represent mean ± SD. Statistically significant differences from control are indicated as follows: ***p < 0.001 and **p < 0.01 (Student’s t test)
Effects of PFOA on ER levels
Since previous studies have reported that PFOA has estrogenic effects (White et al. 2011; Kjeldsen and Bonefeld-Jorgensen 2013), and that MCF-10A cells can be transformed into a malignant phenotype by estrogenic compounds (Hemachandra et al. 2012), we investigated the PFOA effects on ER levels. Western blot analysis with ERα and β antibodies and 17β-estradiol (E2) as a positive control was performed. E2 was found to increase both ERα and ERβ levels after 72 h of exposure, while PFOA had no effects on ER levels (Fig. 4a, b).

Effect of PFOA and 17β-estradiol (E2—positive control) on ERα (a) and ERβ (b) protein levels in MCF-10A breast cells. The cells were exposed to 100 µM PFOA or 10 nM E2 for 72 h. β-tubulin was used as a loading control. Representative blots of three experiments are shown. The results of densitometry analysis are expressed as ER protein band density normalized to the density of β-tubulin bands. Data are reported as mean ± SD of three independent experiments. Statistically significant differences from control are indicated as follows: **p < 0.01 and *p < 0.05 (one-way ANOVA followed by the Tukey–Kramer test)
Hormone receptors involvement in the effects caused by PFOA on MCF-10A proliferation
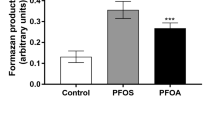
The ER blocker ICI 182,780 was used to examine the role of ER activation on PFOA-induced proliferation. However, the treatment of MCF-10A with ER blocker did not prevent the stimulatory effects of PFOA on cell proliferation (Fig. 5). Since one suggested mode of action for PFOA-induced toxicity is agonism of the nuclear hormone receptor PPAR-α, and activation of the PXR also may occur (Biegel et al. 2001; Lau et al. 2007), we then investigated the involvement of these two receptors. The results showed that the PPARα blocker GW 6471, but not the PXR antagonist sulforaphane was able to prevent MCF-10A proliferation induced by PFOA (Fig. 5).

Involvement of endocrine receptors in the effects induced by PFOA. To determine the role of endocrine receptor, activation cells were preincubated with 100 nM ICI 182,780, 1 µM sulforaphane or 1 µM GW 6471 followed by 100 µM PFOA and the viability was determined by the MTT assay. Data are reported as mean ± SD. Statistically significant differences are indicated as follows: ***p < 0.001 compared with control and ###p < 0.001 compared with PFOA group (two-way ANOVA followed by the Tukey–Kramer test)
Discussion
The breast cancer incidence has globally increased during the last decades (Jemal et al. 2011). Risk factors and mechanistic pathways that can lead to mammary carcinogenesis need to be further elucidated. Recent experimental and epidemiological research suggest an association of the ubiquitously detected environmental contaminants, PFOS and PFOA with tumorigenesis, including breast cancer (Bonefeld-Jorgensen et al. 2011; Lau et al. 2007; Vieira et al. 2013; Wielsoe et al. 2017). However, the causative molecular and biochemical events that may lead to the adverse health outcomes after exposure to these PFAAs are largely undefined.
We recently reported that PFOS promotes MCF-10A proliferation by altering important regulatory cell cycle proteins and accelerating the cell cycle (Pierozan and Karlsson 2018). Despite the structural and many functional similarities, PFOS and PFOA have different toxicological effects (Tsuda 2016). Corroborating with this, the present study revealed that PFOA induces proliferation and acceleration of cell cycle by affecting different regulatory cell cycle proteins. PFOA-induced MCF-10A proliferation by down-regulation of p27 and up-regulation of CDK6, CDK4 and cyclin D levels. Similar to PFOS, PFOA-induced cell migration and invasion, illustrating their capability to induce neoplastic transformation of human normal breast epithelial cells. Unlike PFOS, the ER was not involved in the effects caused by PFOA in MCF-10A cells.
A characteristic feature of malignant tumors is deregulation of the cell cycle, which can occur on many levels, including one or several of the proteins involved in cell cycle control and progression. The progression through the G1–S phase—an important step in tumor development—is regulated by changes in the activity of specific CDKs, with CDK2/CDK4-CDK6 controlling the transition from G1 to S phase (Lundberg and Weinberg 1998). These enzymes, along with their corresponding cyclins D, allow the cell to enter or not in the S phase (Berthet and Kaldis 2007). During G1 phase, the predominant cyclin-CDK complexes are cyclin D-CDK4/6. Here, we found that PFOA increase cyclin D as well as CDK4/6 levels. This could explain the increase in MCF-10A proliferation. Cyclin D is overexpressed in up to 50% of primary breast cancers (Sutherland and Musgrove 2004). Interestingly, an association between overexpression of the cyclin D gene and hormone receptor expression has been observed in breast tumors (Jares et al. 1997; Worsley et al. 1996). These observations support the hypothesis that one of the mechanisms by which endocrine disruptors stimulates breast cancer cell proliferation might be through cyclin D induction.
In addition, CDK4/6 has a pivotal role in the G1–S-phase cell cycle transition in cancer (O’Leary et al. 2016). CDK4 is also found to be highly expressed in aggressive tumors and its expression correlate with poor overall and relapse free survival outcomes as well as poor prognostic features of breast cancer patients, suggesting a central role for this protein in cancer development and progression (Massague 2004). Moreover, up-regulated CDK6 is associated with the development of several types of cancers, and its high expression confers resistant of treatment in breast cancer cells (Alves et al. 2016; Tadesse et al. 2015).
Simultaneous with the increase in cyclin D and CDK4/6 levels, we found a decrease in the levels of the CDK inhibitor p27. Because of their inhibitory activity on cell cycle progression, the inhibitors are considered potential tumor suppressor genes. p27 negatively regulates the G1–S phase progression by binding to and inhibiting cyclin E-CDK2 or cyclin D-CDK4 (Sherr and Roberts 1999). p27 is one of the more recently highlighted cell cycle-related proteins and has the potential to predict outcome in several types of tumors. This protein effectively induces cell cycle arrest and decreases cyclin-CDK activity in breast cancer cell lines (Craig et al. 1997). Low levels, or loss, of p27 expression is a significant predictor of reduced survival, tumor progression and prognosis (Abbas and Dutta 2009; Catzavelos et al. 1997). Moreover, the p27 levels usually decreases during tumor development and progression (Vidal and Koff 2000), and there is considerable evidence that p27 inactivation is fundamental for the development of malignancies (Loda et al. 1997).
Metastasis is the leading cause of cancer-related death in most tumor types. Invasive cancers may spread into surrounding tissue, enter local vasculature, and metastasis to distant sites (McAllister et al. 2011). Cancer cell invasion involves the breaching of tissue barriers by cancer cells, and the subsequent infiltration of these cells throughout the surrounding tissue (McSherry et al. 2007). The first barrier faced by invasive cancer cells is the basement membrane, a dense and rigid matrix. Using a transwell and matrigel assay, we showed that PFOA enhanced the migration and invasion capacity of MCF-10A cells. This effect could be related to the decreased levels of p27, since it is well known that decreased concentration of this inhibitor is implicated in high tumor grade and metastasis in several tumor types including breast carcinomas (Alkarain and Slingerland 2004).
Most of the human breast cancers are initially estrogen dependent (Black et al. 1983). Estrogen-activated ERα regulates the expression of several key cell cycle regulatory genes, such as c-myc, c-fos and cyclin D1 (Prall et al. 1997; van der Burg et al. 1989). On the other hand, studies have reported that ERβ is frequently lost during carcinogenesis, suggesting a role for ERβ as a tumor suppressor (Park et al. 2003; Skliris et al. 2003). In the present study, PFOA had no effect on ERα and ERβ protein expression in MCF-10A cells. Moreover, in contrast to PFOS, the ER blocker ICI 182,780 was not able to prevent the cell proliferation caused by the compound. Instead, the results revealed that the PFOA stimulatory effects on MCF-10A proliferation involve activation of PPARα.
PFOA has previously been shown to activate PPARα, which may be the primary identifiable mode of action for PFOA-induced toxicity (Wolf et al. 2008). PPARα is a transcriptional regulator of lipid and glucose metabolism but also important for diverse functions as keratinocyte differentiation and skin diseases including epidermal and melanoma tumors (Michalik and Wahli 2007; Yang et al. 2006b). Although limited research has investigated the possible link between PPARα and breast cancer, the biology of the gene suggests that it could play a role in the pathology. One compelling hypothesis involves the PPARα-dependent modulation of cell cycle regulatory genes and effects on cell proliferation. According with this, mice fed with the potent PPARα ligand Wy-14 643 show an up-regulation of mRNA and protein for cyclin D1 and CDK4, which agree with our findings. In addition, a transgenic mouse study has suggested that altered PPARα-signaling has the potential to greatly interfere with proper differentiation of the mammary gland (Yang et al. 2006a), which is in line with the finding that PFOA exposure can cause a significant reduction in mammary differentiation in dams (White et al. 2007).
Taken together, PFOA promotes proliferation of MCF-10A cells, induces cell cycle progression by up-regulating the levels of cyclin D1, CDK4/6 and down-regulating the CDK inhibitor p27, through non-estrogenic mechanisms. The compound also increases the metastatic potential of MCF-10A cells by inducing the capability of migration and invasion. This indicates that PFOA is capable of transforming the human normal breast epithelial cell line MCF-10A to a malignant profile. Blocking of PPARα prevented the cell proliferation, suggesting a vital role of this receptor. An overview of the proposed mechanism of PFOA toxicity is depicted in Fig. 6. The detailed mechanisms responsible for the effects of PFOA is unclear, and further studies are needed to shed light on the in vivo effect of PFOA on breast cancer development.

Schematic model of how PFOA may promote cell cycle progression, stimulate cell proliferation and transformation in MCF-10A cells. PFOA acts through PPARα, up-regulating cyclin D1 and CDK4/6 and down-regulating p27, to drive cells into the cell cycle from G1 to S, and promote cell cycle progression
References
Abbas T, Dutta A (2009) p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9(6):400–414. https://doi.org/10.1038/nrc2657
Alkarain A, Slingerland J (2004) Deregulation of p27 by oncogenic signaling and its prognostic significance in breast cancer. Breast Cancer Res 6(1):13–21. https://doi.org/10.1186/bcr722
Alves CL, Elias D, Lyng M et al (2016) High CDK6 protects cells from fulvestrant-mediated apoptosis and is a predictor of resistance to fulvestrant in estrogen receptor-positive metastatic breast cancer. Clin Cancer Res 22(22):5514–5526. https://doi.org/10.1158/1078-0432.CCR-15-1984
Barry V, Winquist A, Steenland K (2013) Perfluorooctanoic acid (PFOA) exposures and incident cancers among adults living near a chemical plant. Environ Health Perspect 121(11–12):1313–1318. https://doi.org/10.1289/ehp.1306615
Bartell SM, Calafat AM, Lyu C, Kato K, Ryan PB, Steenland K (2010) Rate of decline in serum PFOA concentrations after granular activated carbon filtration at two public water systems in Ohio and West Virginia. Environ Health Perspect 118(2):222–228. https://doi.org/10.1289/ehp.0901252
Berthet C, Kaldis P (2007) Cell-specific responses to loss of cyclin-dependent kinases. Oncogene 26(31):4469–4477. https://doi.org/10.1038/sj.onc.1210243
Biegel LB, Hurtt ME, Frame SR, O’Connor JC, Cook JC (2001) Mechanisms of extrahepatic tumor induction by peroxisome proliferators in male CD rats. Toxicol Sci 60(1):44–55
Black R, Prescott R, Bers K, Hawkins A, Stewart H, Forrest P (1983) Tumour cellularity, oestrogen receptors and prognosis in breast cancer. Clin Oncol 9(4):311–318
Bonefeld-Jorgensen EC, Long M, Bossi R et al (2011) Perfluorinated compounds are related to breast cancer risk in Greenlandic Inuit: a case control study. Environ Health 10:88. https://doi.org/10.1186/1476-069X-10-88
Brody JG, Moysich KB, Humblet O, Attfield KR, Beehler GP, Rudel RA (2007) Environmental pollutants and breast cancer: epidemiologic studies. Cancer 109(12 Suppl):2667–2711. https://doi.org/10.1002/cncr.22655
Campeau PM, Foulkes WD, Tischkowitz MD (2008) Hereditary breast cancer: new genetic developments, new therapeutic avenues. Hum Genet 124(1):31–42. https://doi.org/10.1007/s00439-008-0529-1
Catzavelos C, Bhattacharya N, Ung YC et al (1997) Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat Med 3(2):227–230
Emmett EA, Shofer FS, Zhang H, Freeman D, Desai C, Shaw LM (2006) Community exposure to perfluorooctanoate: relationships between serum concentrations and exposure sources. J Occup Environ Med 48(8):759–770. https://doi.org/10.1097/01.jom.0000232486.07658.74
Gullick JW, Bianco C, Normanno N et al (1998) Growth factors and their receptors: a novel approach to the endocrinology of human breast cancer. Women Cancer 1:29–59
Hah YS, Joo HH, Kang YH et al (2014) Cultured human periosteal-derived cells have inducible adipogenic activity and can also differentiate into osteoblasts in a perioxisome proliferator-activated receptor-mediated fashion. Int J Med Sci 16(11):1116–1128 11(
Hemachandra LP, Madhubhani P, Chandrasena R et al (2012) Hops (Humulus lupulus) inhibits oxidative estrogen metabolism and estrogen-induced malignant transformation in human mammary epithelial cells (MCF-10A). Cancer Prev Res (Phila) 5(1):73–81. https://doi.org/10.1158/1940-6207.CAPR-11-0348
Herrick RL, Buckholz J, Biro FM et al (2017) Polyfluoroalkyl substance exposure in the Mid-Ohio River Valley, 1991–2012. Environ Pollut 228:50–60. https://doi.org/10.1016/j.envpol.2017.04.092
Jares P, Rey MJ, Fernandez PL et al (1997) Cyclin D1 and retinoblastoma gene expression in human breast carcinoma: correlation with tumour proliferation and oestrogen receptor status. J Pathol 182(2):160-166. https://10.1002/(SICI)1096-9896(199706)182:2<160::AID-PATH814>3.0.CO;2-2
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61(2):69–90. https://doi.org/10.3322/caac.20107
Jensen AA, Leffers H (2008) Emerging endocrine disrupters: perfluoroalkylated substances. Int J Androl 31(2):161–169. https://doi.org/10.1111/j.1365-2605.2008.00870.x
Kjeldsen LS, Bonefeld-Jorgensen EC (2013) Perfluorinated compounds affect the function of sex hormone receptors. Environ Sci Pollut Res Int 20(11):8031–8044. https://doi.org/10.1007/s11356-013-1753-3
Kortenkamp A (2006) Breast cancer, oestrogens and environmental pollutants: a re-evaluation from a mixture perspective. Int J Androl 29(1):193–198. https://doi.org/10.1111/j.1365-2605.2005.00613.x
Lau C, Anitole K, Hodes C, Lai D, Pfahles-Hutchens A, Seed J (2007) Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicol Sci 99(2):366–394. https://doi.org/10.1093/toxsci/kfm128
Loda M, Cukor B, Tam SW et al (1997) Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med 3(2):231–234
Lundberg AS, Weinberg RA (1998) Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol 18(2):753–761
Maestri L, Negri S, Ferrari M et al (2006) Determination of perfluorooctanoic acid and perfluorooctanesulfonate in human tissues by liquid chromatography/single quadrupole mass spectrometry. Rapid Commun Mass Spectrom 20(18):2728–2734. https://doi.org/10.1002/rcm.2661
Massague J (2004) G1 cell-cycle control and cancer. Nature 432(7015):298–306. https://doi.org/10.1038/nature03094
McAllister SD, Murase R, Christian RT et al (2011) Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res Treat 129(1):37–47. https://doi.org/10.1007/s10549-010-1177-4
McSherry EA, Donatello S, Hopkins AM, McDonnell S (2007) Molecular basis of invasion in breast cancer. Cell Mol Life Sci 64(24):3201–3218. https://doi.org/10.1007/s00018-007-7388-0
Michalik L, Wahli W (2007) Peroxisome proliferator-activated receptors (PPARs) in skin health, repair and disease. Biochim Biophys Acta 1771(8):991–998. https://doi.org/10.1016/j.bbalip.2007.02.004
O’Leary B, Finn RS, Turner NC (2016) Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 13(7):417–430. https://doi.org/10.1038/nrclinonc.2016.26
Park BW, Kim KS, Heo MK et al (2003) Expression of estrogen receptor-beta in normal mammary and tumor tissues: is it protective in breast carcinogenesis? Breast Cancer Res Treat 80(1):79–85. https://doi.org/10.1023/A:1024406223619
Pierozan P, Karlsson O (2018) PFOS induces proliferation, cell-cycle progression, and malignant phenotype in human breast epithelial cells. Arch Toxicol 92(2):705–716. https://doi.org/10.1007/s00204-017-2077-8
Pierozan P, Biasibetti H, Schmitz F et al (2016) Quinolinic acid neurotoxicity: differential roles of astrocytes and microglia via FGF-2-mediated signaling in redox-linked cytoskeletal changes. Biochim Biophys Acta 1863(12):3001–3014. https://doi.org/10.1016/j.bbamcr.2016.09.014
Pozarowski P, Darzynkiewicz Z (2004) Analysis of cell cycle by flow cytometry. Methods Mol Biol 281:301–311. https://doi.org/10.1385/1-59259-811-0:301
Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL (1997) Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem 272(16):10882–10894
Rayne S, Forest K (2009) Perfluoroalkyl sulfonic and carboxylic acids: a critical review of physicochemical properties, levels and patterns in waters and wastewaters, and treatment methods. J Environ Sci Health A Tox Hazard Subst Environ Eng 44(12):1145–1199. https://doi.org/10.1080/10934520903139811
Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13(12):1501–1512
Shin HM, Vieira VM, Ryan PB, Steenland K, Bartell SM (2011) Retrospective exposure estimation and predicted versus observed serum perfluorooctanoic acid concentrations for participants in the C8 Health Project. Environ Health Perspect 119(12):1760–1765. https://doi.org/10.1289/ehp.1103729
Skliris GP, Munot K, Bell SM et al (2003) Reduced expression of oestrogen receptor beta in invasive breast cancer and its re-expression using DNA methyl transferase inhibitors in a cell line model. J Pathol 201(2):213–220. https://doi.org/10.1002/path.1436
Smith PK, Krohn RI, Hermanson GT et al (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150(1):76–85
Sonthithai P, Suriyo T, Thiantanawat A, Watcharasit P, Ruchirawat M, Satayavivad J (2016) Perfluorinated chemicals, PFOS and PFOA, enhance the estrogenic effects of 17beta-estradiol in T47D human breast cancer cells. J Appl Toxicol 36(6):790–801. https://doi.org/10.1002/jat.3210
Soule HD, Maloney TM, Wolman SR et al (1990) Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res 50(18):6075–6086
Steenland K, Fletcher T, Savitz DA (2010) Epidemiologic evidence on the health effects of perfluorooctanoic acid (PFOA). Environ Health Perspect 118(8):1100–1108. https://doi.org/10.1289/ehp.0901827
Sutherland RL, Musgrove EA (2004) Cyclins and breast cancer. J Mammary Gland Biol Neoplasia 9(1):95–104. https://doi.org/10.1023/B:JOMG.0000023591.45568.77
Tadesse S, Yu M, Kumarasiri M, Le BT, Wang S (2015) Targeting CDK6 in cancer: State of the art and new insights. Cell Cycle 14(20):3220–3230. https://doi.org/10.1080/15384101.2015.1084445
Tsuda S (2016) Differential toxicity between perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA). J Toxicol Sci 41(Special):SP27-SP36. https://doi.org/10.2131/jts.41.SP27
van der Burg B, van Selm-Miltenburg AJ, de Laat SW, van Zoelen EJ (1989) Direct effects of estrogen on c-fos and c-myc protooncogene expression and cellular proliferation in human breast cancer cells. Mol Cell Endocrinol 64(2):223–228
Vidal A, Koff A (2000) Cell-cycle inhibitors: three families united by a common cause. Gene 247(1–2):1–15
Vieira VM, Hoffman K, Shin HM, Weinberg JM, Webster TF, Fletcher T (2013) Perfluorooctanoic acid exposure and cancer outcomes in a contaminated community: a geographic analysis. Environ Health Perspect 121(3):318–323. https://doi.org/10.1289/ehp.1205829
White SS, Calafat AM, Kuklenyik Z et al (2007) Gestational PFOA exposure of mice is associated with altered mammary gland development in dams and female offspring. Toxicol Sci 96(1):133–144. https://doi.org/10.1093/toxsci/kfl177
White SS, Fenton SE, Hines EP (2011) Endocrine disrupting properties of perfluorooctanoic acid. J Steroid Biochem Mol Biol 127(1–2):16–26. https://doi.org/10.1016/j.jsbmb.2011.03.011
Wielsoe M, Kern P, Bonefeld-Jorgensen EC (2017) Serum levels of environmental pollutants is a risk factor for breast cancer in Inuit: a case control study. Environ Health 16(1):56. https://doi.org/10.1186/s12940-017-0269-6
Wolf CJ, Takacs ML, Schmid JE, Lau C, Abbott BD (2008) Activation of mouse and human peroxisome proliferator-activated receptor alpha by perfluoroalkyl acids of different functional groups and chain lengths. Toxicol Sci 106(1):162–171. https://doi.org/10.1093/toxsci/kfn166
Worsley SD, Jennings BA, Khalil KH, Mole M, Girling AC (1996) Cyclin D1 amplification and expression in human breast carcinoma: correlation with histological prognostic markers and oestrogen receptor expression. Clin Mol Pathol 49(1):M46–M50
Wrobel AM, Gregoraszczuk EL (2014) Actions of methyl-, propyl- and butylparaben on estrogen receptor-alpha and -beta and the progesterone receptor in MCF-7 cancer cells and non-cancerous MCF-10A cells. Toxicol Lett 230(3):375–381. https://doi.org/10.1016/j.toxlet.2014.08.012
Yang Q, Xie Y, Depierre JW (2000) Effects of peroxisome proliferators on the thymus and spleen of mice. Clin Exp Immunol 122(2):219–226
Yang Q, Kurotani R, Yamada A, Kimura S, Gonzalez FJ (2006a) Peroxisome proliferator-activated receptor alpha activation during pregnancy severely impairs mammary lobuloalveolar development in mice. Endocrinology 147(10):4772–4780. https://doi.org/10.1210/en.2006-0437
Yang Q, Yamada A, Kimura S, Peters JM, Gonzalez FJ (2006b) Alterations in skin and stratified epithelia by constitutively activated PPARalpha. J Invest Dermatol 126(2):374–385. https://doi.org/10.1038/sj.jid.5700056
Zhou J, Liu M, Zhai Y, Xie W (2008) The antiapoptotic role of pregnane X receptor in human colon cancer cells. Mol Endocrinol 22(4):868–880. https://doi.org/10.1210/me.2007-0197
Acknowledgements
The BioVis platform at Uppsala University is gratefully acknowledge for providing the flow cytometry equipment and excellent support.
Funding
This work was supported by the Swedish research council Formas and Carl Tryggers foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pierozan, P., Jerneren, F. & Karlsson, O. Perfluorooctanoic acid (PFOA) exposure promotes proliferation, migration and invasion potential in human breast epithelial cells. Arch Toxicol 92, 1729–1739 (2018). https://doi.org/10.1007/s00204-018-2181-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-018-2181-4