
Abstract
Aims/hypothesis
Increasing brown adipose tissue (BAT) activity is a possible therapeutic strategy to increase energy expenditure and glucose and lipid clearance to ameliorate obesity and associated comorbidities. The thiazolidinedione (TZD) class of glucose-lowering drugs increase BAT browning in preclinical experimental models but whether these actions extend to humans in vivo is unknown. The aim of this study was to determine the effect of pioglitazone treatment on adipocyte browning and adaptive thermogenesis in humans.
Methods
We first examined whether pioglitazone treatment of cultured human primary subacromioclavicular-derived adipocytes induced browning. Then, in a blinded, placebo-controlled, parallel trial, conducted within the Baker Institute clinical research laboratories, 14 lean male participants who were free of cardiometabolic disease were randomised to receive either placebo (lactose; n = 7, age 22 ± 1 years) or pioglitazone (45 mg/day, n = 7, age 21 ± 1 years) for 28 days. Participants were allocated to treatments by Alfred Hospital staff independent from the study via electronic generation of a random number sequence. Researchers conducting trials and analysing data were blind to treatment allocation. The change in cold-stimulated BAT activity, assessed before and after the intervention by [18F]fluorodeoxyglucose uptake via positron emission tomography/computed tomography in upper thoracic and cervical adipose tissue, was the primary outcome measure. Energy expenditure, cardiovascular responses, core temperature, blood metabolites and hormones were measured in response to acute cold exposure along with body composition before and after the intervention.
Results
Pioglitazone significantly increased in vitro browning and adipogenesis of adipocytes. In the clinical trial, cold-induced BAT maximum standardised uptake value was significantly reduced after pioglitazone compared with placebo (−57 ± 6% vs −12 ± 18%, respectively; p < 0.05). BAT total glucose uptake followed a similar but non-significant trend (−50 ± 10% vs −6 ± 24%, respectively; p = 0.097). Pioglitazone increased total and lean body mass compared with placebo (p < 0.05). No other changes between groups were detected.
Conclusions/interpretation
The disparity in the actions of pioglitazone on BAT between preclinical experimental models and our in vivo human trial highlight the imperative to conduct human proof-of-concept studies as early as possible in BAT research programmes aimed at therapeutic development. Our clinical trial findings suggest that reduced BAT activity may contribute to weight gain associated with pioglitazone and other TZDs.
Trial registration
ClinicalTrials.gov NCT02236962
Funding
This work was supported by the Diabetes Australia Research Program and OIS scheme from the Victorian State Government.
Similar content being viewed by others

Introduction
Brown adipose tissue (BAT) is well recognised for its thermogenic (heat-producing) capacity and associated energy expenditure, which distinguish it as a potential anti-obesity therapeutic target [1]. Current research is predicated on BAT’s key function of heat production, which is invoked primarily in response to acute cold exposure through sympathetic nerve pathways. Integral to BAT’s role in thermogenesis is its ability to adapt to chronic cold exposure over weeks and months in a process called adaptive thermogenesis. During this process BAT’s thermogenic capacity increases such that energy expenditure in response to an equivalent stimulus is increased [1]. A recent series of studies demonstrated cold-induced adaptive BAT thermogenesis in humans [2,3,4,5]. Chronic cold exposure is not, however, likely to be tolerated in humans and is associated with increased food intake, therefore is unlikely to be successful as an anti-obesity strategy. A critical step in the development of BAT’s therapeutic potential is identifying a means to pharmacologically induce BAT adaptive thermogenesis in humans. To this end, most investigations in humans have examined the uptake of glucose into BAT using [18F]fluorodeoxyglucose ([18F]FDG) uptake via positron emission tomography/computed tomography (PET/CT) as a surrogate for BAT thermogenesis.
Numerous studies have explored the concept of mimicking the sympathetic nervous system to drive BAT recruitment and activation. In mice, chronic administration of the sympathomimetic ephedrine increased uncoupling protein-1 (UCP-1) content and BAT activity [6]. However, when the pan-β-adrenergic receptor agonist, ephedrine, was administered for 4 weeks to humans, BAT glucose uptake decreased [7]. This effect, likely due to downregulation of β1/2-adrenergic receptor signalling, highlighted distinct species differences between humans and rodents and the necessity to identify alternate approaches. Numerous mechanisms that putatively induce adipose tissue browning have been identified in preclinical research [8], yet none have been investigated in humans.
By activating peroxisome proliferator-activated receptor γ (PPARγ) in adipocytes, thiazolidinediones (TZDs) promote adipocyte differentiation, adipogenesis and browning [9]. TZDs are also putatively the only exogenous agents that have been conclusively demonstrated to induce browning of adipose tissue, although this has yet to be determined in humans in vivo [10]. TZDs are not activators of thermogenesis per se, but can induce browning and therefore potentially permit greater responsiveness to an activating stimulus (i.e. adrenergic activation).
Mouse models with adipose-specific PPARγ ablation demonstrate impaired BAT development [11] and mice treated with PPARγ agonists have profound expansion of BAT mass [12]. TZDs also induce browning in human adipocyte culture models [13] and rosiglitazone is used almost universally to promote differentiation and browning of human cultured brown adipocytes [13,14,15,16,17]. Rosiglitazone and, to a lesser extent, pioglitazone were widely prescribed for type 2 diabetes until safety concerns were raised (primarily with rosiglitazone [18]), leaving pioglitazone as the predominantly prescribed TZD worldwide [19]. The aim of this study was therefore to investigate the effect of pioglitazone on browning and adaptive thermogenesis in human brown/beige adipose tissue through both a novel cell culture model and a human clinical trial.
Methods
Overview
This study consisted of two parts: (1) a human primary cell culture study to determine whether pioglitazone could induce adipogenesis and browning of primary adipocytes derived from subacromioclavicular adipose biopsies and (2) upon determination that pioglitazone induced browning in vitro, a clinical trial to examine whether prolonged (4 weeks) treatment of humans with pioglitazone would increase cold-stimulated BAT activity, assessed by glucose uptake using [18F]FDG–PET/CT. Both studies were approved by the Alfred Health Ethics Committee and the cell culture study was also approved by Ramsay Health Human Research Ethics Committee and performed in accordance with the Declaration of Helsinki, Seventh Revision, 2013. All participants provided written, informed consent.
Human cell culture study
Participants
Twenty-six individuals (14 women, 12 men; 54 ± 2 years) admitted for shoulder reconstructive surgery donated subacromial adipose tissue. This region corresponds with [18F]FDG uptake during cold exposure [20, 21] and would therefore be expected to contain a mixture of adipocyte and pre-adipocyte subtypes, including brown/beige adipocytes, and is directly relevant to informing our subsequent clinical trial in which upper thoracic and cervical adipose tissue is the primary region of interest.
Tissue isolation, culture and experimental procedures
Tissue was placed in ice-cold sterile DMEM and transported to the laboratory. Within 60 min, the tissue was dissected free from connective tissue, minced into ~1 mm pieces and then cells were extracted and cultured as described [15].
Cells derived from 10–15 individuals were combined to create aggregate lines to produce sufficient material for repeated experiments. Cells were plated into 12 or 24 well assay plates and assayed to determine the effect of the TZDs pioglitazone and rosiglitazone on markers of browning. After 2 weeks of differentiation, cells were fixed in 10% (wt/vol.) neutral-buffered formalin and then stained with Oil Red O (surrogate indicator of general adipogenesis). The cells were assessed for lipolytic rate (basal and stimulated with 1 μmol/l isoprenaline [known as isoproterenol in the USA] for 2 h to assess general adipose function) or UCP-1 protein content was determined (indicator of cell browning) as described [15]. Treatments included vehicle (DMSO), 1 μmol/l rosiglitazone or 1 μmol/l pioglitazone. TZD concentrations were selected based on studies of single clinical doses of these drugs (15–45 mg) which resulted in comparable plasma concentrations (0.5 μmol, l–2 μmol/l) [22, 23], and previous human primary adipocyte experiments [15]. Individual experiments were conducted with three or more technical replicates/treatment condition. The mean of technical replicates within experiments was considered n = 1. Within each experiment, the fold change for TZDs relative to vehicle was determined and these values presented and used for statistical comparisons. Individual experiments were repeated four or five times. Treatment conditions and samples were not randomised or blinded during these experiments. Any experiments excluded from final data analyses were done on the basis of poor quality cultures (overt cell death on visual inspection or lack of differentiation due to mistreatment or infection) and/or experimental procedures and data analysis (errors evident from internal controls, poor quality western blotting, sample contamination).
Clinical study
Participants
Seeking proof-of-concept, and as the first study of its type in humans, we examined lean, healthy participants. Fifteen male participants (aged 19–30 years, BMI ≤ 25 kg/m2, free from cardiovascular disease and diabetes, unmedicated, sedentary, non-smokers) participated in this study, conducted between June 2015 and July 2016. One participant withdrew from the trial prior to completion due to non-compliance with the protocol. Therefore, 14 participants completed the study and were included in data analyses described.
Study design
The trial was of a randomised, blinded, placebo-controlled and parallel-study design. Participants received either pioglitazone (45 mg/day, n = 7) or placebo (lactose, n = 7) for 28 days. The pioglitazone dose is the highest dose regularly prescribed to lower blood glucose in the treatment of type 2 diabetes and was chosen to maximise treatment effects. Randomisation was conducted using Microsoft Excel (v2013, Southbank, VIC, Australia) to generate a random order sequence.
Participants visited the research facilities on three occasions. During their first visit, a general medical screen was conducted and baseline physical (height, weight, waist:hip ratio, BP, body composition via dual-energy x-ray absorptiometry) and biochemical (glucose, insulin, HbA1c, lipids) characteristics were measured. The second and third visits were scheduled before and after the intervention and included assessment of cold-stimulated BAT glucose uptake via [18F]FDG–PET/CT imaging, whole-body energy expenditure (indirect calorimetry) and blood metabolites and hormones (glucose, NEFA, noradrenaline, thyroid hormones [T3, T4, thyroid-stimulating hormone (TSH)]). HOMA-IR was calculated as [insulin (pmol/l) × glucose (mmol/l)]/22.5. Upon completion of visit 2, dosing commenced with participants taking oral pioglitazone (45 mg) or placebo daily for 28 days. Participants then returned to the laboratory (visit 3) the following day to repeat the protocol conducted at visit 2, including body composition reassessment.
Experimental protocol for BAT activation trial
Initial screening involved clinical history and examination by a physician [7].
Before and after the 28 days of intervention on the evening prior to both experimental days (visits 2 and 3), participants consumed a standardised meal (3180 kJ; 84% carbohydrate, 13% protein, 3% fat) between 18:00 and 22:00 h. Upon arrival (07:30–08:00 h) after an overnight fast and abstention from vigorous exercise, caffeine, smoking and alcohol consumption for ≥2 days prior, participants voided and changed into hospital scrubs and socks. They consumed a telemetric pill for recording core temperature (HQ, Palmetto, FL, USA) and an antecubital venous cannula was inserted. Brachial BP (Philips Suresigns VS3; Philips Medical Systems, Andover, MA, USA) was measured every 15 min and heart rate (Cortemp; HQ) was continuously recorded. Participants then rested in a supine position for 2 h while covered with two blankets to ensure thermoneutrality. Laboratory temperature was maintained at 22–24°C.
After the participants had rested for 2 h, energy expenditure was measured via indirect calorimetry (TrueOne 2400; Parvo-Medics, East Sandy, UT, USA) and a blood sample was taken. Participants then underwent standardised cold exposure via a temperature-controlled, water-perfused vest and a similarly perfused blanket placed under the exposed feet and legs (Polar Products, Stow, OH, USA). The initial water temperature was 14°C and the temperature was then lowered by 1°C every 5 min until participants reported signs of shivering. The temperature was then raised by 1–2°C until shivering ceased and was maintained thereafter. For each participant, garment water temperature was recorded during visit 2 and repeated at visit 3. Final perfusate temperatures were not different between groups (10.8 ± 0.8°C, placebo; 11.8 ± 0.8°C, pioglitazone). Blood samples were taken at 30, 60 and 90 min after cold exposure for subsequent analyses (described below). Participants were injected with [18F]FDG for BAT glucose uptake assessment via PET/CT 60 min after induction of cooling. Energy expenditure was measured 60–90 min after cold exposure. Finally, participants were taken to the Alfred Hospital Department of Nuclear Medicine and PET, where PET/CT imaging was completed 60 min after injection of [18F]FDG.
Indirect calorimetry
Energy expenditure was measured as previously described [7, 24].
PET/CT imaging
PET/CT imaging and analyses were conducted as previously described [7, 24], except PET/CT image acquisition and reconstruction was carried out using a GE Discovery 710 PET/CT scanner (GE, Fairfield, CT, USA) and analysis was performed using MIM software v6.6 (MIM Software, Cleveland, OH, USA).
Image analysis focused on the region defined from the base of the fourth cervical vertebra to the top of the third thoracic vertebra. Adipose tissue was identified in this region by CT radiodensity ranging from −190 to −10 Hounsfield units [25]. Within these regions, a range of predetermined standardised uptake values (SUVs) on PET images was used to define BAT glucose uptake. The primary results described are based on an SUV threshold of >1.5. Additionally, since the literature on measurement of BAT activity in humans via [18F]FDG–PET/CT has included a range of SUV thresholds, we also conducted the same analyses with thresholds of 0 (no threshold), >1.0 and >2.0.
BAT glucose uptake was reported within the regions described above as the maximum SUV (SUVmax) and BAT total glucose uptake (BAT TGU; SUVmean multiplied by total volume of voxels registering above the designated SUV threshold). SUVmax was defined as the highest registered SUV of any voxel within the defined regions and SUVmean as the mean SUV of all voxels within a defined area. Both SUVmax and SUVmean were corrected for lean body mass according to recent recommendations [25].
Biochemical analyses
Where indicated, plasma was centrifuged and frozen for analyses or whole blood was collected in an appropriate preservative and measured immediately as described [7, 24].
Outcome measures
The primary outcome measure was change in BAT glucose uptake between groups. Secondary outcome measures comprised changes in basal and cold-stimulated energy expenditure, body composition, circulating hormones, lipids and metabolites.
Power calculations
The sample size was based on minimising the number of healthy volunteers exposed to ionising radiation. Assuming a standard deviation for change in BAT glucose uptake of 18%, the sample size of seven per group powered the study to detect a minimum difference between groups for a change of 30% (power 80%, α = 0.05). Given the actual difference in SUVmax of 45% (pioglitazone −57% vs placebo −12%), the study was adequately powered.
Statistical analyses
All data were tested for normality. Data from cell experiments were not normally distributed, therefore treatments compared using a non-parametric Kruskal–Wallis ANOVA and Conover–Iman test. For the clinical trial, physical characteristics were normally distributed and compared between pioglitazone and placebo groups using an unpaired two-tailed Student’s t test. The change (post-intervention − pre-intervention) between the pioglitazone and placebo groups (BAT SUVmax and BAT TGU, indirect calorimetry, cardiovascular and metabolite data) were normally distributed and therefore compared using ANCOVA. Covariates included baseline values for each variable and the change in maximum daily temperature was averaged over 4 weeks prior to each experimental visit. The effect of cold on energy expenditure was assessed by repeated measures ANOVA. Analyses were conducted using SPSS (v22; IBM, St Leonards, NSW, Australia), Microsoft Excel (v2013) and Stata (v14; StataCorp, College Station, TX, USA). Data are expressed as mean ± SEM with p < 0.05 considered significant.
Results
Human cell culture study
Compared with vehicle treatment, rosiglitazone and pioglitazone significantly increased adipogenesis, lipolysis and UCP-1 protein in human primary subacromioclavicular pre-adipocytes (p < 0.05, Fig. 1). The effects of rosiglitazone and pioglitazone were similar for all measurements (p > 0.99).

The adipogenic, lipolytic and browning effects of rosiglitazone (Rosi) and pioglitazone (Pio) were compared with those of vehicle (Veh) in human primary subacromioclavicular pre-adipocytes. (a) Oil Red O imaging and quantification (n = 4) (light microscopy, magnification ×4). (b) Isoprenaline-stimulated lipolysis (n = 5). (c) UCP-1 protein (n = 4). Data are expressed as the fold change vs vehicle-treated cells (Veh = 1) and are presented as mean ± SEM. *p < 0.05 vs Veh
Clinical trial
Table 1 shows baseline participant characteristics. Groups were similar for all variables.
BAT glucose uptake
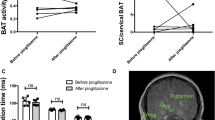
Cold-induced BAT SUVmax was significantly reduced (−57 ± 6%) after pioglitazone compared with placebo treatment (−12 ± 18%) (Fig. 2a; placebo, pre-intervention 22.2 ± 6.5, post-intervention 18.4 ± 6.0; pioglitazone, pre-intervention 21.5 ± 3.1, post-intervention 9.4 ± 0.6; p < 0.05). BAT TGU displayed a similar but non-significant trend (Fig. 2b; placebo, pre-intervention 520 ± 167, post-intervention 412 ± 162; pioglitazone, pre-intervention 496 ± 105, post-intervention 217 ± 39; p = 0.097).

Individual cold-induced SUVmax (n = 7/group) (a) and BAT TGU (n = 7/group) (b) in supraclavicular brown/beige adipose tissue before (Pre) and after (Post) 28 days of treatment with placebo or pioglitazone (45 mg/day). Data are presented as mean ± SEM. *p < 0.05 for change (Post – Pre) in BAT SUVmax between groups
Past studies examining BAT activity/glucose uptake in humans using [18F]FDG–PET/CT have employed variable SUV thresholds to determine BAT glucose uptake, therefore we also quantified BAT TGU based on no SUV threshold and thresholds of >1.0 and >2.0. These calculations, together with the above data using an SUV threshold of >1.5, are presented in electronic supplementary material (ESM) Table 1 and corresponding CT radiodensity data are shown in ESM Table 2. Of note, while none of the differences achieved significance, for BAT TGU, as threshold values increased the difference between pioglitazone and placebo groups increased such that it approached significance, suggesting a trend towards decreased BAT glucose uptake in the pioglitazone group (SUV >2.0, p = 0.075). Interestingly, albeit to a lesser degree, differences in CT radiodensity for pioglitazone vs placebo also approached statistical significance, but in the direction of ‘total’ adipose tissue (no SUV thresholding, p = 0.091) rather than ‘BAT’ (thresholding for SUV of >1.0, 1.5 or 2.0), indicative of increased total adipose lipid content in the pioglitazone group.
There was no difference in the average maximum daily temperature for the 4 week period preceding measurement of BAT glucose uptake either within or between groups (placebo, pre-intervention 18.2 ± 1.6°C and post-intervention 19.8 ± 2.1°C; pioglitazone, pre-intervention 18.4 ± 1.6°C and post-intervention 21.2 ± 1.7°C).
Cold-stimulated [18F]FDG uptake for several tissues are shown in ESM Table 3. Pioglitazone treatment did not alter [18F]FDG uptake in any of the tissues examined.
Body composition
Body composition at baseline and in response to the intervention is presented in Table 2. The change in total mass (placebo, −0.03 ± 0.5 kg; pioglitazone, 1.34 ± 0.4 kg; p = 0.03) and lean mass (placebo, −0.19 ± 0.3 kg; pioglitazone, 1.04 ± 0.3 kg; p = 0.02) were significantly greater after pioglitazone than after placebo (Table 2; p < 0.05). Fat mass, visceral adipose tissue and bone mineral content were unchanged.
Energy expenditure
Basal and cold-stimulated energy expenditure and respiratory exchange ratio (RER) remained unchanged after chronic pioglitazone treatment (Table 3).
Cardiovascular responses, core temperature and blood metabolites
The changes in systolic BP, heart rate, core temperature, blood glucose, plasma noradrenaline and plasma NEFA in response to acute cold exposure are shown in Fig. 3 before and after the intervention for both groups. There was no difference in the response to cold exposure between groups. Additionally, basal concentrations of circulating hormones, metabolites and lipids were not different between groups (Table 1) and were not affected by pioglitazone treatment (Table 4).

Change (Δ, Post − Pre) in systolic blood pressure (SBP) (a), heart rate (HR) (b), core temperature (c), blood glucose (d), plasma noradrenaline (NA) (e) and plasma NEFA (f) from prior to cold exposure (0 min) to the mean of values taken between 60 and 90 min after cold exposure, before (Pre) and after (Post) 28 days of treatment with placebo or pioglitazone (45 mg/day). Data are presented as mean ± SEM. Placebo: n = 7 for all variables, except Post NEFA (n = 5); pioglitazone: n = 7 for all variables. White bars, placebo; black bars, pioglitazone
Discussion
This study is the first to investigate the effect of chronic TZD treatment on human BAT activity, assessed by cold-stimulated glucose ([18F]FDG) uptake in vivo. Contrary to our hypothesis, previous preclinical evidence and data from our in vitro experiments in human primary adipocytes, cold-induced BAT glucose uptake decreased in response to pioglitazone treatment. This suggests that reduced BAT activity may contribute to the weight gain associated with pioglitazone and other TZDs [26] previously attributed to proliferation and differentiation of subcutaneous white adipocytes, increased insulin sensitivity and fluid retention [9, 27]. In this context, pioglitazone-induced BAT dysregulation could be counteractive and potentially worsen obesity-related comorbidities.
A large body of work supports the notion that TZD treatment recruits BAT. Previous TZD studies in patients with type 2 diabetes [14], obese rats and mice [28] and cultured human subcutaneous adipocytes [13, 15] showed that chronic treatment with a TZD increases browning in brown and/or white adipose tissues. Our human cell culture experiments involving pre-adipocytes derived from a region known to contain brown/beige adipocyte precursors corroborated this. We therefore hypothesised that pioglitazone treatment in vivo would increase cold-induced BAT glucose uptake.
While TZDs induce adipogenesis and browning [29], the increase in browning may not directly translate to an enhanced response to thermogenic stimuli. Adaptive thermogenesis elicited by chronic cold not only involves increased BAT browning, but also development of the BAT neurovascular network via sympathetic nerve branching and angiogenesis which increases nutrient delivery and heat dissipation [30]. These processes, which are essential for maximal BAT adrenergic responsiveness, may not be stimulated by pioglitazone. This is, however, speculative and ideally adipose biopsies from relevant depots would be taken to examine molecular changes. This was not possible here, but will inform future in vivo human studies.
While several lines of evidence suggest that TZDs promote BAT thermogenic adaptation, one laboratory demonstrated that rosiglitazone reduced sympathetic activation of BAT in rodents [31, 32]. BAT was found to be recruited by rosiglitazone independently of sympathetic tone, but maximal UCP-1 expression required adrenergic ‘priming’ [33]. In their studies, the adrenergic ‘prime’ was reduced by lowered sympathetic drive linked to chronic reduction in thyroid signalling within BAT [31]. Although it was not possible to measure global or BAT-specific sympathetic tone in this study, we did not observe changes in either circulating catecholamines or T3, T4 and TSH in response to pioglitazone. This does not rule out TZD-induced reduction in sympathetic outflow to BAT but suggests that a mechanism alternative to sympathetic–thyroid axis downregulation may be responsible for our observation. Pioglitazone reportedly inhibits β3-adrenergic receptor mRNA transcription in brown adipocytes [34]; the effect of TZDs on thermogenic potential is therefore complex and may depend on a balance between sympathetic outflow, catecholamine turnover, thyroid hormone production, expression of β3-adrenergic receptors and UCP-1, and other thermogenic regulatory factors. Further studies are required to elucidate the mechanisms involved in TZD-induced regulation of BAT recruitment and activity.
Despite reduction in cold-stimulated BAT glucose uptake in response to pioglitazone, neither energy expenditure (basal and cold-stimulated) nor RER were altered by pioglitazone. This was not unexpected because potential alterations in BAT energy expenditure and fuel selection would be small and masked by tissues that make larger contributions towards whole-body energy expenditure. This result is consistent with human BAT (in contrast to observations in rodent models) making a relatively minor contribution towards total energy expenditure. More direct and sensitive methods are required to assess any contribution of human BAT to substrate utilisation.
Several clinical studies have reported body weight increases after chronic TZD treatment (>12 weeks) and that this increase in mass corresponds to increased fat and lean mass [26]. The relatively short duration of the present study and participant characteristics likely explain the lack of increased fat mass. Given the trend towards increased total adipose lipid content, based on decreased radiodensity in the pioglitazone group (ESM Table 2), it is likely that longer treatment in these healthy individuals would result in a measurable increase in fat mass. The observed increase in lean mass is likely attributable to fluid retention, a well-known side-effect of TZD treatment associated with water retention in the kidneys that, in comparison with other mechanisms of increased mass, occurs rapidly, independent of disease status [27]. Increased muscle glycogen synthesis and its associated water content may increase lean mass. This, however, has only been reported in insulin-resistant animals and humans [35]. In the current study, whole-body insulin sensitivity (HOMA-IR and muscle [18F]FDG uptake) was unchanged by pioglitazone. Nevertheless, like fat mass, it remains unknown whether this mechanism would become apparent after several months of TZD treatment in insulin-sensitive lean individuals.
Clinical implications
Prior to 2008, rosiglitazone and pioglitazone were widely prescribed. Data from The Health Improvement Network (THIN) primary care database in the UK suggest that these drugs accounted for over 3 prescriptions per 1000 person-years, with rosiglitazone making more than double the contribution of pioglitazone [36]. Within 18 months of publication of the meta-analysis questioning the cardiovascular safety of rosiglitazone [18], the number of rosiglitazone prescriptions halved and the number of pioglitazone prescriptions doubled [36]. Overall, while the TZD prescription rate is lower now than in 2007, currently, 10% of all prescriptions for patients with type 2 diabetes in the THIN cohort are prescribed a TZD [37] and pioglitazone retains its position at the frontline of diabetes management [19, 37].
Obesity, type 2 diabetes and BAT dysfunction are all linked [24, 38, 39]. While evidence supporting the association with BAT dysfunction in humans is associative only, the possibility that BAT dysfunction contributes to obesity and metabolic dysregulation cannot be dismissed. Accordingly, based on the evidence presented here, it cannot be excluded that TZD treatment promotes weight gain through inhibition of BAT thermogenesis. On balance, the weight of evidence from clinical trials suggests that TZD treatment improves metabolic control in patients with type 2 diabetes. Nevertheless, in individuals with adequate BAT function, inhibition by TZD treatment may exacerbate obesity and impair metabolic homeostasis. Alternatively, because the patients for whom TZDs are prescribed are known to exhibit poor BAT function and are rarely exposed to BAT-activating stimuli, it could be argued that TZDs cannot contribute to further disease progression through this mechanism.
Methodological strengths and limitations
Our in vitro study methodology provided a robust system by which to validate the cell autonomous browning capability of pioglitazone in a model closely representing the in vivo target tissue. Nevertheless, this highlights the difficulty in interrogating the in vivo response to TZDs and translating the effects of any drug from basic to clinical science. Several central and peripheral mechanisms are likely to be involved, which, despite presumably driving brown/beige adipocyte browning, integrate to decrease thermogenic responsiveness of these cells. Of note, because we were not able to biopsy the relevant tissue depots, we cannot confirm whether pioglitazone induced browning of fat cells in the supraclavicular and cervical depots.
It is possible that the increase in body mass (attributable entirely to lean mass) confounded the observation of reduced BAT glucose uptake in the pioglitazone group. However, the increase in tracer volume of distribution because of the very small (~2%) increase in lean mass is accounted for by lean mass correction in the calculation of SUV. Furthermore, the >50% decrease in BAT SUVmax and BAT TGU occurred in conjunction with no change in SUVmax in any other tissues and was of far greater magnitude than the change in lean mass. This evidence supports our contention that pioglitazone decreased cold-stimulated BAT glucose uptake.
Although [18F]FDG–PET/CT is informative with respect to BAT activity, it does not provide a comprehensive assessment of BAT distribution and metabolic activity [40]. During BAT activation in response to cold exposure, the primary substrates utilised for BAT thermogenesis are intracellular lipids [40, 41]. Secondary fuels are extracted from the circulation by BAT, most likely in preferential order of NEFA followed by glucose [1]. Because glucose is not the primary substrate, BAT activity based on [18F]FDG uptake may underestimate and potentially miss components of BAT energy metabolism under certain physiological conditions. While [18F]FDG–PET-based assessments of human BAT activity in response to acute [20, 21, 38, 42] and chronic [2, 4] cold exposure have reflected the expected outcomes based on rodent experiments, this study indicates that there are species differences for other adaptive stimuli. Although we have shown that pioglitazone reduced cold-stimulated BAT glucose uptake, we cannot definitively claim that fat oxidative capacity also decreased. Nevertheless, previous acute cold interventions in similar participants resulted in parallel kinetics between fatty acid and glucose PET tracers [42], thus it is likely that our findings are relevant for BAT thermogenic activity. Our study design minimised the impact of individual and non-BAT [18F]FDG uptake variability by conducting pre- and post-imaging on all participants, tightly controlling laboratory conditions and maximising statistical and experimental rigor by including a placebo-treated group. Nevertheless, further studies are warranted to interrogate the impact of TZDs on human BAT function.
Conclusion
For the first time, we examined BAT glucose uptake in response to chronic TZD treatment in humans in vivo. Contrary to our hypothesis, cold-induced BAT glucose uptake was reduced. The mechanisms explaining this are currently unknown but are likely to be complex, involving a combination of peripheral organs and altered regulation of sympathetic output. These data may have implications for the use of TZDs in certain individuals with diabetes and highlight the importance of conducting human in vivo studies in parallel with preclinical investigations.
Change history
08 December 2017
The baseline insulin data given in Table 1 for the placebo group were incorrectly reported as 51 ± 10 pmol/l instead of 48 ± 10 pmol/l. This mistake also impacts on data reported in Table 4.
Abbreviations
- BAT:
-
Brown adipose tissue
- BAT TGU:
-
BAT total glucose uptake
- [18F]FDG:
-
[18F]Fluorodeoxyglucose
- PET/CT:
-
Positron emission tomography/computed tomography
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- RER:
-
Respiratory exchange ratio
- SUV:
-
Standardised uptake value
- TSH:
-
Thyroid-stimulating hormone
- TZD:
-
Thiazolidinedione
- UCP-1:
-
Uncoupling protein-1
References
Cannon B, Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359
Blondin DP, Labbe SM, Tingelstad HC et al (2014) Increased brown adipose tissue oxidative capacity in cold-acclimated humans. J Clin Endocrinol Metab 99:E438–E446
Lee P, Smith S, Linderman J et al (2014) Temperature-acclimated brown adipose tissue modulates insulin sensitivity in humans. Diabetes 63:3686–3698
van der Lans AA, Hoeks J, Brans B et al (2013) Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest 123:3395–4403
Yoneshiro T, Aita S, Matsushita M et al (2013) Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest 123:3404–3408
Young P, Wilson S, Arch JR (1984) Prolonged beta-adrenoceptor stimulation increases the amount of GDP-binding protein in brown adipose tissue mitochondria. Life Sci 34:1111–1117
Carey AL, Pajtak R, Formosa MF et al (2015) Chronic ephedrine administration decreases brown adipose tissue activity in a randomised controlled human trial: implications for obesity. Diabetologia 58:1045–1054
Villarroya F, Cereijo R, Villarroya J, Giralt M (2017) Brown adipose tissue as a secretory organ. Nat Rev Endocrinol 13:26–35
Soccio RE, Chen ER, Lazar MA (2014) Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 20:573–591
Nedergaard J, Cannon B (2014) The browning of white adipose tissue: some burning issues. Cell Metab 20:396–407
Imai T, Takakuwa R, Marchand S et al (2004) Peroxisome proliferator-activated receptor γ is required in mature white and brown adipocytes for their survival in the mouse. Proc Natl Acad Sci U S A 101:4543–4547
Tai TA, Jennermann C, Brown KK et al (1996) Activation of the nuclear receptor peroxisome proliferator-activated receptor gamma promotes brown adipocyte differentiation. J Biol Chem 271:29909–29914
Digby JE, Montague CT, Sewter CP et al (1998) Thiazolidinedione exposure increases the expression of uncoupling protein 1 in cultured human preadipocytes. Diabetes 47:138–141
Bogacka I, Xie H, Bray GA, Smith SR (2005) Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 54:1392–1399
Carey AL, Vorlander C, Reddy-Luthmoodoo M et al (2014) Reduced UCP-1 content in in vitro differentiated beige/brite adipocytes derived from preadipocytes of human subcutaneous white adipose tissues in obesity. PLoS One 9:e91997
Elabd C, Chiellini C, Carmona M et al (2009) Human multipotent adipose-derived stem cells differentiate into functional brown adipocytes. Stem Cells 27:2753–2760
Jespersen NZ, Larsen TJ, Peijs L et al (2013) A classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metab 17:798–805
Nissen SE, Wolski K (2007) Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 356:2457–2471
Schernthaner G, Currie CJ, Schernthaner GH (2013) Do we still need pioglitazone for the treatment of type 2 diabetes? A risk-benefit critique in 2013. Diabetes Care 36(Suppl 2):S155–S161
Cypess AM, Chen YC, Sze C et al (2012) Cold but not sympathomimetics activates human brown adipose tissue in vivo. Proc Natl Acad Sci U S A 109:10001–10005
Saito M, Okamatsu-Ogura Y, Matsushita M et al (2009) High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 58:1526–1531
Sherwin CM, Ding L, Kaplan J, Spigarelli MG, Vinks AA (2011) Optimal study design for pioglitazone in septic pediatric patients. J Pharmacokinet Pharmacodyn 38:433–447
Kadam R, Bourne D, Kompella U, Aquilante C (2013) Effect of cytochrome P450 2C8*3 on the population pharmacokinetics of pioglitazone in healthy caucasian volunteers. Biol Pharm Bull 36:245–251
Carey AL, Formosa MF, Van Every B et al (2013) Ephedrine activates brown adipose tissue in lean but not obese humans. Diabetologia 56:147–155
Chen KY, Cypess AM, Laughlin MR et al (2016) Brown adipose reporting criteria in imaging STudies (BARCIST 1.0): recommendations for standardized FDG-PET/CT experiments in humans. Cell Metab 24:210–222
Domecq JP, Prutsky G, Leppin A et al (2015) Clinical review: drugs commonly associated with weight change: a systematic review and meta-analysis. J Clin Endocrinol Metab 100:363–370
Beltowski J, Rachanczyk J, Wlodarczyk M (2013) Thiazolidinedione-induced fluid retention: recent insights into the molecular mechanisms. PPAR Res 2013:628628
Kelly LJ, Vicario PP, Thompson GM et al (1998) Peroxisome proliferator-activated receptors gamma and alpha mediate in vivo regulation of uncoupling protein (UCP-1, UCP-2, UCP-3) gene expression. Endocrinology 139:4920–4927
Tontonoz P, Spiegelman BM (2008) Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 77:289–312
Cao Y (2007) Angiogenesis modulates adipogenesis and obesity. J Clin Invest 117:2362–2368
Festuccia WT, Blanchard PG, Oliveira TB et al (2012) PPARγ activation attenuates cold-induced upregulation of thyroid status and brown adipose tissue PGC-1α and D2. Am J Physiol Regul Integr Comp Physiol 303:R1277–R1285
Festuccia WT, Oztezcan S, Laplante M et al (2008) Peroxisome proliferator-activated receptor-γ-mediated positive energy balance in the rat is associated with reduced sympathetic drive to adipose tissues and thyroid status. Endocrinology 149:2121–2130
Festuccia WT, Blanchard PG, Richard D, Deshaies Y (2010) Basal adrenergic tone is required for maximal stimulation of rat brown adipose tissue UCP1 expression by chronic PPAR-γ activation. Am J Physiol Regul Integr Comp Physiol 299:R159–R167
Bakopanos E, Silva JE (2000) Thiazolidinediones inhibit the expression of beta3-adrenergic receptors at a transcriptional level. Diabetes 49:2108–2115
Boden G, Cheung P, Mozzoli M, Fried SK (2003) Effect of thiazolidinediones on glucose and fatty acid metabolism in patients with type 2 diabetes. Metabolism 52:753–759
Leal I, Romio SA, Schuemie M et al (2013) Prescribing pattern of glucose lowering drugs in the United Kingdom in the last decade: a focus on the effects of safety warnings about rosiglitazone. Br J Clin Pharmacol 75:861–868
Sharma M, Nazareth I, Petersen I (2016) Trends in incidence, prevalence and prescribing in type 2 diabetes mellitus between 2000 and 2013 in primary care: a retrospective cohort study. BMJ Open 6:e010210
van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM et al (2009) Cold-activated brown adipose tissue in healthy men. N Engl J Med 360:1500–1508
Hanssen MJ, Hoeks J, Brans B et al (2015) Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat Med 21:863–865
Cypess AM, Haft CR, Laughlin MR, Hu HH (2014) Brown fat in humans: consensus points and experimental guidelines. Cell Metab 20:408–415
Cypess AM, Kahn CR (2010) Brown fat as a therapy for obesity and diabetes. Curr Opin Endocrinol Diabetes Obes 17:143–149
Ouellet V, Labbe SM, Blondin DP et al (2012) Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest 122:545–552
Acknowledgements
We thank C. Despott (Department of Nuclear Medicine and PET) and S. Phillips (Human Neurotransmitters Laboratory) for technical assistance, and the research participants for their time and interest in our study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding authors on reasonable request.
Funding
This work was supported by the Diabetes Australia Research Program and OIS scheme from the Victorian State Government. BAK (Senior Principal Research Fellowship) and GWL (Senior Research Fellowship) hold National Health and Medical Research Council Fellowships.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
RKCL, MFF, DAB, MJA, SAB, KSY, TWB, MHC, SJD, BAK and ALC contributed to study conception and design. RKCL, MFF, NE, DAB, MJA, SAB, SN, NDC, ALG, ATR, KSY, TWB, GWL, MHC, SJD and ALC contributed to data collection. RKCL, NE, ALG, KSY, TWB, GWL, MHC, BAK and ALC contributed to data analysis. All authors contributed to discussion, writing and critique of the manuscript and gave approval for the final version to be published. ALC is the guarantor of this work.
Additional information
A correction to this article is available online at https://doi.org/10.1007/s00125-017-4514-x.
Electronic supplementary material
ESM Tables
(PDF 183 kb)
Rights and permissions
About this article

Cite this article
Loh, R.K.C., Formosa, M.F., Eikelis, N. et al. Pioglitazone reduces cold-induced brown fat glucose uptake despite induction of browning in cultured human adipocytes: a randomised, controlled trial in humans. Diabetologia 61, 220–230 (2018). https://doi.org/10.1007/s00125-017-4479-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-017-4479-9