
Abstract
In parallel with the growing diabetes pandemic, there is an increasing burden of micro- and macrovascular complications, occurring in the majority of patients. The identification of a number of synergistic accelerators of disease, providing therapeutic pathways, has stabilised the incidence of complications in most western nations. However, the primary instigators of diabetic complications and, thus, prevention strategies, remain elusive. This has necessitated a refocus on natural history studies, where tissue and plasma samples are sequentially taken to determine when and how disease initiates. In addition, recent Phase III trials, wherein the pleiotropic effects of compounds were arguably as beneficial as their glucose-lowering capacity in slowing the progression of complications, have identified knowledge gaps. Recently the influence of other widely recognised pathological pathways, such as mitochondrial production of reactive oxygen species, has been challenged, highlighting the need for a diverse and robust global research effort to ascertain viable therapeutic targets. Technological advances, such as -omics, high-resolution imaging and computational modelling, are providing opportunities for strengthening and re-evaluating research findings. Newer areas such as epigenetics, energetics and the increasing scrutiny of our synergistic inhabitants, the microbiota, also offer novel targets as biomarkers. Ultimately, however, this field requires concerted lobbying to support all facets of diabetes research.
Similar content being viewed by others

Introduction
The greatest part of the morbidity and mortality associated with diabetes results from chronic vascular complications. These are broadly divided into micro- and macrovascular complications [1]. Microvascular complications include diabetic kidney disease (DKD), retinopathy and peripheral neuropathy. Development of microvascular disease, particularly DKD, is a potent risk factor for macrovascular disease, namely atherosclerosis, myocardial infarction, heart failure and cerebrovascular events. Overall, the rates of diabetic complications in developed nations have stabilised as the result of improved management of risk factors such as hyperglycaemia, hypertension and dyslipidaemia [2]. However, this has been offset by the increasing absolute number of new diabetes cases, particularly in developing nations. Hence, the actual numbers of individuals affected by diabetes complications is still increasing.
The quest for novel therapies to prevent and/or treat diabetes complications remains paramount. We can certainly stabilise disease, but we cannot prevent it. While we should continue to treat the major risk factors, we must not ignore new pathways that could provide blockbuster therapies. Furthermore, we must ask ourselves whether we really understand how complications develop and progress over time and whether our new therapies are hitting the target. It is well appreciated that only one discovered therapy in around 100 will ever reach clinical trials for diabetes complications and the odds of discovering a therapy that can prevent or reverse disease are even lower. Over the past decade, there have been clinical trial failures but also some heartening successes.
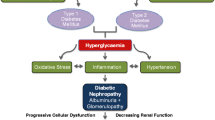
Significant overlap exists between mechanisms contributing to macrovascular and microvascular complications in both major forms of diabetes (Fig. 1) and it is appreciated that they may have a common pathological instigator [1]. The proportion of patients affected by complications after diabetes of long duration is commonly consistent in both type 1 and type 2 diabetes and genetic forms of diabetes such as MODY, supporting this postulate. Furthermore, given the number of comorbidities seen with type 2 diabetes, often before ‘official’ diabetes diagnosis, it is conceivable that this explains the perception of more rapid onset of complications in these individuals. Interestingly, there is also evidence that early progression to complications may be clinically ‘silent’, as previously described for type 1 diabetes [3], making temporal patterns of disease development difficult to map.

Schematic representation of the pathways to diabetes complications discussed in this review and how they fit into current paradigms. Susceptibility to diabetes complications is driven by a combination of environmental factors (e.g. diet, lifestyle, microbiota, pathogen exposure) and genetic programming. With diabetes onset, many changes occur that are thought to be pathological, including involvement of pathways that have been explored recently and are highlighted in this review. These include the following: changes to the microbiome, potentially affecting substrate delivery and utilisation, gastrointestinal inflammation and permeability, release of intestinal toxins, neuroendocrine signalling and the immune system; aberrant energy utilisation, substrate delivery and nutrient flux, which can alter the metabolic pathways utilised by tissues affected by complications; mitochondrial dysfunction in the form of mitochondrial fission and fusion, decreased biogenesis, aberrant energy utilisation and delivery and, potentially, ROS generation; epigenetic changes, which alter the regulation of genes associated with pathological pathways. All these factors are likely to be exacerbated by the presence of comorbidities (obesity, raised blood pressure, dyslipidaemia, endothelial dysfunction), initiating a downstream cascade of interacting pathways ultimately resulting in microvascular and macrovascular complications. These pathways include post-translational modifications (AGE formation, oxidation of proteins and lipids and ER stress), inflammation and immune dysregulation (increases in inflammatory cytokines, chemoattractant molecules and ultimately immune cell infiltration), ROS production, and gene expression and transcription. Current therapeutic strategies include intensive blood glucose control and treatment of comorbidities, with the former appearing to be more effective early in disease development. The paucity of therapies that actually prevent or reverse complications once they are established remains one of the major challenges posed by the global diabetes pandemic. ECM, extracellular matrix; ER, endoplasmic reticulum
Finally, this review serves as a reminder that there is significant value in research time spent convincing governments and funding bodies to ensure equitable research expenditure per affected individual, comparable with diseases such as AIDS and cancer.
New slants on old theories: ‘know thine enemy’
Glucose control
Since the DCCT [4] and the UK Prospective Diabetes Study (UKPDS [5]) showed that early intensive glucose control reduced the risk for complications, this has been a major clinical goal in diabetic individuals. These trials supported the findings of earlier in vivo preclinical studies, leading to the ‘metabolic memory hypothesis’ wherein aggressive early control of blood glucose alone is postulated to dampen vascular stressors, reducing the risk of vascular complications [6]. The mechanisms behind metabolic memory are still debated, however, with both epigenetic programming (discussed below) and AGEs being potential mediators [7]. In line with this, surrogate measures of AGEs, such as skin auto-fluorescence, are increasingly being evaluated as predictors of both glucose control and risk for diabetic complications [8].
Studies have shown a clear temporal bias attached to the benefits of strict glycaemic control on complications (Fig. 2). First, studies such as Action to Control Cardiovascular Risk in Diabetes (ACCORD) [9] and Action in Diabetes and Vascular Disease (ADVANCE) [10], in older individuals with longer duration of diabetes and at least one risk factor for macrovascular disease, lend support to intensive glycaemic control decreasing the incidence of microvascular disease. However, in these studies, intensive glycaemic control did not reduce the risk of cardiovascular events or death, suggesting that these individuals had reached a checkpoint in the disease process that could not be influenced by moderating glycaemic control. Identifying this checkpoint would be a worthwhile endeavour. By contrast, renin–angiotensin system (RAS) blockade did show benefits on macrovascular endpoints in ADVANCE [11] and the UKPDS. It remains to be determined whether this was via direct blood-pressure-lowering or pleiotropic effects of this class of agents. Indeed, a large meta-analysis of antihypertensive therapy in DKD has shown a lack of effect of blood-pressure lowering on mortality [12].

Flow chart showing therapeutic strategies in diabetes complications at different phases of the research pipeline. The chart shows known pathways in the pathogenesis of diabetes complications followed by the therapeutic strategies and clinical trials targeting that specific pathway. Arrows with dashed borders represent checkpoints in the translational pipeline that have not yet been reached. ACCORD, Action to Control Cardiovascular Risk in Diabetes; ACTION, Aminoguanidine Clinical Trial In Overt Type 2 diabetic Nephropathy; ALTITUDE, Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints; ARTS-DN, Mineralocorticoid Receptor Antagonist Tolerability Study-Diabetic Nephropathy; ASK-1, Apoptosis signal-regulating kinase 1; BEACON, Breast Cancer Outcomes With NKTR-102; CAS9, CRISPR-associated protein 9; CRISPR, clustered regularly interspaced short palindromic repeats; DIVINE, Dialysis Infection and Vitamin D in New England; EMPA-REG, Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes trial; ETDRS, Early Treatment Diabetic Retinopathy Study; FIELD, Fenofibrate Intervention and Event Lowering in Diabetes; GI, gastrointestinal; GLO1, glyoxalase 1; HDAC, histone deacetylase; HDACi, HDAC inhibitor; HOPE, Heart Outcomes Prevention Evaluation; Jnk-1, c-Jun-N terminal protein kinase; JPAD, Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes; KO, knockout; LEADER, Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results; MAPK, mitogen activated protein kinase; mito, mitochondrial; Nox, NADPH oxidase; P38, p38 mitogen activated protein kinase; POPADAD, The prevention of progression of arterial disease and diabetes; PREDIMED, Prevention with a Mediterranean Diet; RAGE, receptor for AGEs; SGLT2i, SGLT2 inhibitor
At the opposite end of the disease spectrum (i.e. young persons with diabetes, in whom optimal glycaemic control is often difficult achieve) the findings are also puzzling. Adolescents with type 1 diabetes and markers reflecting the onset of renal and cardiovascular disease (elevations in urinary albumin excretion within a normal range, abnormal cardiac autonomic function and increases in aortic intimal–medial thickening) do not have increased HbA1c compared with matched (age, sex, diabetes duration) individuals without these changes [13, 14]. In fact, in young individuals with type 1 diabetes with and without evidence of early vascular complications it is difficult to ascertain differences in most conventionally held risk factors (blood pressure, dyslipidaemia, obesity), suggesting that complications can develop and progress in the absence of most known risk factors. Indeed, in adolescence, increases in albumin excretion within the normal range defines a group of individuals with type 1 diabetes having increased risk for renal and macrovascular disease. The Adolescents with Diabetes Intervention Trial (AdDIT) is currently testing the efficacy of RAS blockade and statin therapy in decreasing markers of progressive renal and macrovascular disease in the absence of systemic hypertension or dyslipidaemia [15]. Interestingly, although the DCCT/Epidemiology of Diabetes Interventions and Complications Trial (EDIC) showed that strict glycaemic control for a short period in young adults with type 1 diabetes (13–39 years of age) delayed complications by approximately 6 years, other studies have shown that RAS blockade has no such effect in the same age group [16].
Poor glycaemic control in individuals with early-onset type 2 diabetes (<30 years of age) confers greater risk for developing complications than age-matched individuals with type 1 diabetes or those with adult-onset type 2 diabetes. This is not surprising given that these young individuals with type 2 diabetes would have the highest number of comorbidities (obesity, dyslipidaemia, hypertension) for the development of complications and likely potent genetic or socioeconomic drivers, given the higher incidence in indigenous communities. Taken with the data from older individuals and from adolescents with type 1 diabetes, one could postulate a common underlying pathological mechanism for complications independent of age and the type of diabetes, which can be accelerated by the presence of comorbidities. It remains debatable, however, as to whether any of these comorbidities in isolation lead to complications.
Glycaemic variability
The view that more frequent or greater fluctuations in blood glucose concentrations (glycaemic variability) are more harmful than sustained but stable hyperglycaemia is another area of active research [17]. This variability is postulated to reflect declining beta cell function but is likely far more complex given changes in renal glucose excretion, gluconeogenesis and impaired hypoglycaemic awareness. In any case, it appears that the markers used to assess glycaemic variability are key to understanding this area [18]. Greater pre- and postprandial glucose excursions or HbA1c variability over time do predict increased risk for cardiovascular events. Similarly, in elderly individuals with type 2 diabetes, long-term variability in fasting glucose concentrations is more strongly associated with cardiovascular disease (CVD) mortality and all-cause mortality than the actual blood glucose concentrations at any time [18]. Numerous in vivo and in vitro preclinical studies have also demonstrated that glycaemic excursions result in activation of pathways known to be pathological mediators of complications such as advanced glycation, monocyte adhesion and vascular lesions in the absence of chronic hyperglycaemia [18]. However, there remains a paucity of clinical research to support the notion of glucose variability being a major pathological contributor to diabetes complications.
Next generation glucose-lowering therapies
Promising reductions in composite cardiovascular endpoints and progressive kidney disease in individuals with type 2 diabetes and existing complications or elevated risk for CVD (Fig. 2) have been demonstrated in recent Phase III clinical trials such as the Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results (LEADER) [19], Semaglutide Cardiovascular Outcomes Study (SUSTAIN 6) and the Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes trial (EMPA-REG) [20]. These non-inferiority ‘safety’ studies used the antihyperglycaemic agents liraglutide (glucagon-like peptide [GLP-1] analogue), semaglutide (GLP-1 analogue) and empagliflozin (sodium/glucose cotransporter 2 [SGLT2] inhibitor), respectively. It is important to note that each of these studies were based upon preclinical research performed decades before and that without exception these agents have pleiotropic effects that cannot be separated from their hypoglycaemic actions. For example, the SGLT2 inhibitor empagliflozin significantly lowers systolic blood pressure, changes glomerular filtration rate and facilitates weight loss [20], while GLP-1 analogues decrease body weight and improve lipid profiles, making it impossible to attribute the benefits of these agents to glucose lowering alone. The first-line hypoglycaemic agent metformin has also been reported to have pleiotropic actions; recently it was found to have the ability to increase enterocyte production of GLP-1 in both humans and experimental models [21].
Of course, insulin has potent effects on the incidence of diabetes complications. It will be interesting to see how the advent of systems that provide more consistent pulsatile insulin infusions as well as smarter insulins, affects the incidence of chronic complications, potentially shedding new light on pathways leading to complications.
Energy generation and metabolic flux of nutrients: a newcomer?
Energy generation
There is increasing evidence that organs which are highly metabolic are most at risk for the development of complications [1, 22]. The oxygen-dependent generation of ATP by our cellular power plants, the mitochondria, provides the fuel for most metabolic processes. In the vasculature and smooth muscle cells, however, a large proportion of ATP is generated via glycolytic pathways, often anaerobically. These are intricately balanced systems wherein nutrients such as glucose, fatty acids, amino acids and ketones are processed and, in highly metabolic tissues such as kidneys and the heart [23], are then transferred to the mitochondria to maximise ATP production. The ATP synthesised within the mitochondria by oxidative phosphorylation is then shuttled to various cellular locations to enable metabolism.
Mitochondrial dysfunction, including altered networking (i.e. mitochondrial fission and fusion), decreased biogenesis, altered turnover by cellular autophagy and excess generation of reactive oxygen species (ROS) [24], is evident at many sites of diabetes complications, identified first in preclinical models and later in humans [19, 25]. Ultimately, these morphological and functional changes alter the amount of ATP available in cells. Therapies thought to improve mitochondrial function (Fig. 2), including coenzyme Q10 (CoQ10) [26,27,28], mitoubiquinone mesylate (MitoQ) [29] and the peptide d-Arg-2′,6′-dimethyltyrosine-Lys-Phe-NH2 (SS-31), have beneficial effects on kidney function and fibrosis in models of experimental diabetes [30] and obesity [31].
Nutrient flux
In diabetes, nutrient delivery for energy production is altered by chronic hyperglycaemia, dyslipidaemia and ketosis. Renal studies show increased cellular flux of nutrients early in disease, which later declines, suggesting temporal changes with disease progression [24, 32]. This may explain why glucose-lowering agents are more effective when given earlier in disease. It is not known, however, whether this occurs in all organs affected by complications. Recent metabolomic flux studies in mice given a single oral gavage of isotopic substrate, such as glucose or fatty acids (palmitate-BSA), have supported the postulate that nutrient flux into energy generation pathways occurs at sites of diabetes complications [32]. Unfortunately, these flux studies, as performed so far, may not provide much information beyond that reported in previous work using static measurements, given that they are single-oral-administration studies in which tissue and blood measurements are performed in fasted mice without steady-state infusions of metabolites and lacking control for differences in organ blood flow. However, these studies provide the foundation for more research in this complex area of nutrient transport.
Altering the substrate delivery to organs by manipulation of nutrient transporters has provided further insight into the role of metabolism in diabetic complications. Changing the expression of glucose transporters such as GLUT1 at sites such as the kidney and heart has given mixed results [1]. However, tissue-specific blockade of the insulin-sensitive glucose transporter GLUT4 in the heart and kidney podocytes induced cardiomyopathy and renal disease, respectively [1].
Since dyslipidaemia is prevalent in diabetes and its treatment is aimed at lowering the risk for CVD, the metabolic flux of fatty acids at sites of diabetes complications has recently been investigated [33]. It is evident that high energy consumers such as the kidney and heart rely on sustained fatty acid delivery for normal function. This delivery is impaired in diabetes, causing tissue injury. Paradoxically, the lipid-lowering agent fenofibrate increases fatty acid oxidation in diabetic kidneys in concert with renoprotection. However, fenofibrate does not consistently prevent complications at all sites, appearing to be less effective against macrovascular disease in Phase III clinical studies (Fig. 2; Fenofibrate Intervention and Event Lowering in Diabetes [FIELD]).
Controversies in ROS generation
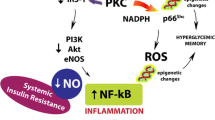
Excess production of mitochondrial superoxide as a master upstream mediator formed the basis of a widely held hypothesis in diabetes complications [21]. Mitochondrial superoxide generation was thought to result in diabetes complications by generating AGEs, increasing hexosamine and polyol pathway flux and activating protein kinase C. This supposition was based primarily on findings from in vitro studies and subsequent discoveries have led to this view being questioned. Several studies have shown that diabetes complications can occur in the absence of excess, or in the presence of decreased, mitochondrial superoxide generation [34]. In the diabetic kidney, increases in superoxide align with improvements in renal function [34]. However, in some preclinical studies, therapies targeting mitochondrial function (e.g. with SS-31, CoQ10 and MitoQ) reduced cellular ROS generation at some sites (Fig. 2). Although the issue of mitochondrial ROS remains contentious, there is evidence to show that mitochondrial function and excess cellular energy production are critical mediators of diabetes complications.
Clinically, the failure of several antioxidants to reduce CVD events in Phase III trials such as Heart Outcomes Prevention Evaluation (HOPE, Fig. 2) has also challenged the view that excess ROS is a mediator of diabetes complications [35]. Other cellular ROS-generating pathways, such as cytosolic NADPH oxidases (Figs 1, 2), tissue-specific complexes which generate superoxide to act as a second messengers, are also implicated in the pathogenesis of diabetic micro- and macrovascular disease [35]. A number of recent studies have shown that blockade or knockdown of the NADPH oxidase isoforms, nox 1 and nox 4, found at most sites of diabetes complications, can prevent renal disease and atherosclerosis. Agents that target both these isoforms simultaneously are in Phase II clinical trials for DKD (ClinicalTrial.gov registration no. NCT02010242).
Genetic susceptibility: it’s in your genes and you can’t change it
Evidence to support heritability as a major driver of vascular complications is found in the familial clustering of DKD, diabetic retinopathy and atherosclerosis [36,37,38,39]. However, given the diversity and interplay of genes, identifying the major players has proven a mammoth task. Inadequate sample size, polygenicity, gene–environment interactions and the added complexity of post-transcriptional regulation have all impeded this search. Furthermore, the clinical characteristics used to define susceptible groups are often highly variable and some, such as albuminuria, can regress in some individuals. More-specific analyses of subsets of diabetic individuals, such as targeted study of those who are rapid progressors to renal impairment, as assessed by GFR [40], have greatly enhanced understanding in this area. There have also been some successes by international consortia, such as Genetic of Kidneys in Diabetes (GoKIND), Genetics of Nephrology—an International Effort (GENIE) and others, in overcoming sample size limitations.
More recently, a novel means of studying the heritability of complications using genome-wide association studies (GWAS) integrated with a targeted functional approach has been applied to genetic loci that approached, but did not reach, significance. Here, networks of gene transcripts within specific pathways can be interrogated by gene promoter modelling [41]. Indeed, it is likely that there will be more integration of -omics datasets in the future to better identify important checkpoints and nodes in the disease process. Other novel approaches to study heritability of diabetes complications currently in their infancy include zebrafish models, the Collaborative Cross gene mine (http://sysgen.org/TheCollaborativeCross/CC.html) and organoid cultures from diseased tissues.
Gene expression: epigenetics and non-coding RNAs
Epigenetic changes, defined as reversible changes that are heritable, affecting gene expression without altering the DNA sequence [42], may explain the proportion of heritable risk for vascular complications not accounted for by GWAS and candidate gene studies. Epigenetic changes include DNA methylation and histone and chromatin modifications (methylation, acetylation, ubiquitination, citrullination, phosphorylation and sumoylation). Non-coding RNAs, such as microRNAs (miRNAs), as well as short and long non-coding RNAs, add an additional level of gene regulation. Indeed, some miRNAs are altered by diabetes and factors previously implicated in diabetes complications (Table 1; [42,43,44]).
Although still a burgeoning field, the stability of elements such as miRNAs in biological fluids makes epigenetics a powerful area for biomarker discovery. A study of salivary DNA from individuals with type 2 diabetes (with or without end-stage renal disease) identified 187 differentially methylated gene targets, of which ~21% corresponded with those identified by GWAS or transcription profiling [45]. In another study, individuals with type 1 diabetes and DKD showed differential methylation in UNC13B, also previously implicated by GWAS studies in the pathogenesis of DKD [46]. Other studies in individuals with type 1 diabetes have also shown differential methylation of genes that influence mitochondrial function in individuals with complications.
In vitro studies have been integral to teasing out epigenetic regulation of pathways activated by hyperglycaemia. At the cellular level, both chronic and transient high glucose exposure leads to sustained expression of various genes, supporting epigenetics as a major driver of metabolic memory. However, care must be taken to accurately represent the diabetic milieu and results must be interpreted cautiously. As an example, Brennan et al [47] have postulated that the exposure time to high glucose concentrations in cell models is not sufficient to reproduce the epigenetic changes that develop over many years in diabetic individuals. However, the robust concordance between epigenetic modifications identified in diabetic individuals and in mouse models of DKD is interesting.
There is also a paucity of data examining gene-specific epigenetic modifications at sites of diabetes complications in humans. One study in nephrectomy tissue identified methylation profiles in individuals with DKD in putative enhancer regions of fibrosis-associated genes, not seen in individuals without DKD [48]. The major limitation of most study designs, however, is that the data represent a single temporal snapshot of disease, making it difficult to ascertain exactly when in the disease process a particular mediator is actually pathological. This could limit our understanding of how genes and other pathways interact at various sites, stressing the need for longitudinal studies of complications in human tissues, beginning much earlier in disease.
Environmental interactions: diet, bugs and the immune system
With obesity a major comorbidity and increased nutrient flux at sites postulated as a pathogenic mediator, it is no surprise that diet has gained attention as a means to alter the development and progression of diabetes complications [1]. However, aside from the obvious benefits of weight loss in overweight or obese individuals, restriction of dietary protein in the context of DKD is the only hard recommendation seen in most clinical guidelines (www2.kidney.org/professionals/KDOQI/guideline_diabetes/guide5.htm, accessed 2 May 2017). Preclinical studies have demonstrated the efficacy of various dietary interventions, such as ketogenic diets, energy restriction and intermittent fasting, in slowing the progression of complications but these remain to be substantiated clinically. To date, the only diet that has conclusively conferred protection against cardiovascular events in diabetic individuals is the Mediterranean diet [49]. Compellingly, researchers showed that after a median follow-up of 4.8 years participants on the Mediterranean diet had a relative risk reduction for macrovascular events of ~30% compared with the control diet group. Hence, it is possible that dietary intervention can be utilised to combat complications and that substantiation and refinement of the clinical guidelines for nutrition in diabetes could be a worthy research endeavour.
The microbiome has become an area of intensive research effort over the past decade, with findings suggesting a role in susceptibility to various human diseases ranging from obesity, diabetes and colitis to disorders on the autism spectrum [50]. Many seminal rodent studies show that changes in the microbiome contribute to both obesity [50] and type 2 diabetes, supported by human studies [50, 51]. Generation of metabolites, release of (or permeability to) intestinal toxins and neuroendocrine and immune dysregulation are postulated as mechanisms by which the microbiota could contribute to diabetes complications (Fig. 1). Some enticing preclinical studies demonstrate slowing of cognitive decline and oxidative stress after 8 weeks of probiotic supplementation [52]. However, the interplay between gut microbiota and sites vulnerable to diabetic vascular complications remains to be experimentally untangled.
Not surprisingly, the immune system is dysregulated by diabetes, where chronic tissue infiltration by leucocytes and sustained production of inflammatory mediators and cytokines are commonly seen at sites of complications [1]. Although a very important component of disease progression in diabetes complications, the complexity of this system and its systemic integration within our bodies demands careful targeting so as not to destabilise this balance. However, targeting of molecules such as apoptosis signal-regulating kinase 1, currently under investigation in Phase II clinical trials for DKD, will provide further insight in this area (Fig. 2).
Conclusions
Despite decades of research into the pathogenesis of diabetes complications, it is disappointing that the pathological pathways that initiate disease remain to be fully elucidated. We have discovered plenty of accelerators and since these have been therapeutically targeted, there is stabilisation of disease. Nevertheless, the absolute numbers of individuals with diabetes complications continue to grow. As research continues, we should be mindful of the temporal progression of complications and the possibility that they may begin earlier than first postulated. Also, when examining the successful interventions influencing human health and disease, it is not surprising to find that their foundations lie decades earlier, in discovery-based science. These discoveries were then translated to clinical research by other pioneers until finally, through work by researchers at the clinical coalface, they became best practice. This pipeline shows the diversity of researchers required at each level. So, advocate for research, to governments and anyone else who will listen, to preserve and expand this diversity of research towards a common goal, to prevent, reverse and treat diabetic complications.
Abbreviations
- ADVANCE:
-
Action in Diabetes and Vascular Disease
- CoQ10:
-
Coenzyme Q10
- CVD:
-
Cardiovascular disease
- DKD:
-
Diabetic kidney disease
- GLP-1:
-
Glucagon-like peptide 1
- GWAS:
-
Genome-wide association study
- miRNA:
-
microRNA
- MitoQ:
-
Mitoubiquinone mesylate
- RAS:
-
Renin–angiotensin system
- ROS:
-
Reactive oxygen species
- SGLT2:
-
Sodium/glucose cotransporter 2
- SS-31:
-
d-Arg-2′,6′-dimethyltyrosine-Lys-Phe-NH2
- UKPDS:
-
UK Prospective Diabetes Study
References
Forbes JM, Cooper ME (2013) Mechanisms of diabetic complications. Physiol Rev 93:137–188
Geiss LS, Wang J, Cheng YJ et al (2014) Prevalence and incidence trends for diagnosed diabetes among adults aged 20 to 79 years, United States, 1980-2012. JAMA 312:1218–1226
Schultz CJ, Konopelska-Bahu T, Dalton RN et al (1999) Microalbuminuria prevalence varies with age, sex, and puberty in children with type 1 diabetes followed from diagnosis in a longitudinal study. Oxford Regional Prospective Study Group. Diabetes Care 22:495–502
The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
UK Prospective Diabetes Study (UKPDS) Group (1998) Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 352:854–865
Ceriello A (2012) The emerging challenge in diabetes: the “metabolic memory”. Vasc Pharmacol 57:133–138
Chilelli NC, Burlina S, Lapolla A (2013) AGEs, rather than hyperglycemia, are responsible for microvascular complications in diabetes: a “glycoxidation-centric” point of view. Nutr Metab Cardiovasc Dis 23:913–919
Gerrits EG, Lutgers HL, Kleefstra N et al (2008) Skin autofluorescence: a tool to identify type 2 diabetic patients at risk for developing microvascular complications. Diabetes Care 31:517–521
Group AtCCRiDS (2008) Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008:2545–2559
Group AC (2008) Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008:2560–2572
Zoungas S, Chalmers J, Neal B et al (2014) Follow-up of blood-pressure lowering and glucose control in type 2 diabetes. N Engl J Med 371:1392–1406
Palmer SC, Mavridis D, Navarese E et al (2015) Comparative efficacy and safety of blood pressure-lowering agents in adults with diabetes and kidney disease: a network meta-analysis. Lancet 385:2047–2056
Cho YH, Craig ME, Davis EA et al (2015) Cardiac autonomic dysfunction is associated with high-risk albumin-to-creatinine ratio in young adolescents with type 1 diabetes in AdDIT (adolescent type 1 diabetes cardio-renal interventional trial). Diabetes Care 38:676–681
Maftei O, Pena AS, Sullivan T et al (2014) Early atherosclerosis relates to urinary albumin excretion and cardiovascular risk factors in adolescents with type 1 diabetes: Adolescent type 1 Diabetes cardio-renal Intervention Trial (AdDIT). Diabetes Care 37:3069–3075
Marcovecchio ML, Woodside J, Jones T et al (2014) Adolescent type 1 diabetes cardio-renal intervention trial (AdDIT): urinary screening and baseline biochemical and cardiovascular assessments. Diabetes Care 37:805–813
Mauer M, Zinman B, Gardiner R, et al. (2009) Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med 361
Bergenstal RM (2015) Glycemic variability and diabetes complications: does it matter? Simply put, there are better glycemic markers! Diabetes Care 38:1615–1621
Ceriello A, Kilpatrick ES (2013) Glycemic variability: both sides of the story. Diabetes Care 36:S272–S275
Correia LC, Latado A, Porzsolt F (2016) Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 375:1798
Heerspink HJ, Perkins BA, Fitchett DH, Husain M, Cherney DZ (2016) Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 134:752–772
McCreight LJ, Bailey CJ, Pearson ER (2016) Metformin and the gastrointestinal tract. Diabetologia 59:426–435
Brownlee M (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54:1615–1625
Wang Z, Ying Z, Bosy-Westphal A et al (2010) Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr 92:1369–1377
Coughlan MT, Nguyen TV, Penfold SA et al (2016) Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin Sci 130:711–720
Hallan S, Sharma K (2016) The role of mitochondria in diabetic kidney disease. Curr Diab Rep 16:61
Dugan LL, You YH, Ali SS et al (2013) AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest 123:4888–4899
Persson MF, Franzen S, Catrina SB et al (2012) Coenzyme Q10 prevents GDP-sensitive mitochondrial uncoupling, glomerular hyperfiltration and proteinuria in kidneys from db/db mice as a model of type 2 diabetes. Diabetologia 55:1535–1543
Sourris KC, Harcourt BE, Tang PH et al (2012) Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic Biol Med 52:716–723
Chacko BK, Reily C, Srivastava A et al (2010) Prevention of diabetic nephropathy in Ins2+/˗AkitaJ mice by the mitochondria-targeted therapy MitoQ. Biochem J 432:9–19
Hou Y, Li S, Wu M et al (2016) Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am J Physiol Ren Physiol 310:F547–F559
Szeto HH, Liu S, Soong Y, Alam N, Prusky GT, Seshan SV (2016) Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int 90:997–1011
Sas KM, Kayampilly P, Byun J et al (2016) Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 1:e86976
Stadler K, Goldberg IJ, Susztak K (2015) The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr Diab Rep 15:40
Coughlan MT, Sharma K (2016) Challenging the dogma of mitochondrial reactive oxygen species overproduction in diabetic kidney disease. Kidney Int 90:272–279
Forbes JM, Coughlan MT, Cooper ME (2008) Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 57:1446–1454
The Diabetes Control and Complications Trial Research Group (1997) Clustering of long-term complications in families with diabetes in the Diabetes Control and Complications Trial. Diabetes 46: 1829–1839
Langefeld CD, Beck SR, Bowden DW, Rich SS, Wagenknecht LE, Freedman BI (2004) Heritability of GFR and albuminuria in Caucasians with type 2 diabetes mellitus. Am J Kidney Dis 43:796–800
Hietala K, Forsblom C, Summanen P, Groop P-H, on behalf of the FinnDiane Study Group (2008) Heritability of proliferative diabetic retinopathy. Diabetes 57:2176–2180
Ma RCW (2016) Genetics of cardiovascular and renal complications in diabetes. J Diabetes Investig 7:139–154
Krolewski AS (2015) Progressive renal decline: the new paradigm of diabetic nephropathy in type 1 diabetes. Diabetes Care 38:954–962
Martini S, Nair V, Patel SR et al (2013) From single nucleotide polymorphism to transcriptional mechanism: a model for FRMD3 in diabetic nephropathy. Diabetes 62:2605–2612
Kato M, Natarajan R (2014) Diabetic nephropathy—emerging epigenetic mechanisms. Nat Rev Nephrol 10:517–530
Beltrami C, Angelini TG, Emanueli C (2015) Noncoding RNAs in diabetes vascular complications. J Mol Cell Cardiol 89:42–50
Reddy MA, Das S, Zhuo C et al (2016) Regulation of vascular smooth muscle cell dysfunction under diabetic conditions by miR-504. Arterioscler Thromb Vasc Biol 36:864–873
Sapienza C, Lee J, Powell J et al (2011) DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics 6:20–28
Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA (2010) Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genet 3:1
Brennan EP, Ehrich M, OʼDonovan H et al (2010) DNA methylation profiling in cell models of diabetic nephropathy. Epigenetics 5:396–401
Ko Y-A, Mohtat D, Suzuki M et al (2013) Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol 14:R108
Estruch R, Ros E, Salas-Salvadó J et al (2013) Primary prevention of cardiovascular disease with a Mediterranean diet. N Engl J Med 368:1279–1290
Griffin JL, Wang X, Stanley E (2015) Does our gut microbiome predict cardiovascular risk? A review of the evidence from metabolomics. Circ Cardiovasc Genet 8:187–191
Turnbaugh PJ, Hamady M, Yatsunenko T et al (2009) A core gut microbiome in obese and lean twins. Nature 457:480–484
Davari S, Talaei SA, Alaei H, Salami M (2013) Probiotics treatment improves diabetes-induced impairment of synaptic activity and cognitive function: behavioral and electrophysiological proofs for microbiome–gut–brain axis. Neuroscience 240:287–296
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
JMF was supported by Fellowships from the National Health and Medical Research Council of Australia (NH&MRC; 1004503; 1102935) and the Mater Foundation. AKF was supported by an Australian Postgraduate Award and the Frank Clair scholarship.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
Both authors were responsible for drafting the article and revising it critically for intellectual content. Both authors approved the version to be published.
Electronic supplementary material
ESM Downloadable slideset
(PPTX 484 kb)
Rights and permissions
About this article

Cite this article
Forbes, J.M., Fotheringham, A.K. Vascular complications in diabetes: old messages, new thoughts. Diabetologia 60, 2129–2138 (2017). https://doi.org/10.1007/s00125-017-4360-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-017-4360-x