
ABSTRACT
Aims/hypothesis
Diabetes mellitus is characterised by beta cell loss and alpha cell expansion. Analogues of glucagon-like peptide-1 (GLP-1) are used therapeutically to antagonise these processes; thus, we hypothesised that the related cell cycle regulators retinoblastoma protein (Rb) and p107 were involved in GLP-1 action.
Methods
We used small interfering RNA and adenoviruses to manipulate Rb and p107 expression in insulinoma and alpha-TC cell lines. In vivo we examined pancreas-specific Rb knockout, whole-body p107 knockout and Rb/p107 double-knockout mice.
Results
Rb, but not p107, was downregulated in response to the GLP-1 analogue, exendin-4, in both alpha and beta cells. Intriguingly, this resulted in opposite outcomes of cell cycle arrest in alpha cells but proliferation in beta cells. Overexpression of Rb in alpha and beta cells abolished or attenuated the effects of exendin-4 supporting the important role of Rb in GLP-1 modulation of cell cycling. Similarly, in vivo, Rb, but not p107, deficiency was required for the beta cell proliferative response to exendin-4. Consistent with this finding, Rb, but not p107, was suppressed in islets from humans with diabetes, suggesting the importance of Rb regulation for the compensatory proliferation that occurs under insulin resistant conditions. Finally, while p107 alone did not have an essential role in islet homeostasis, when combined with Rb deletion, its absence potentiated apoptosis of both alpha and beta cells resulting in glucose intolerance and diminished islet mass with ageing.
Conclusions/interpretation
We found a central role of Rb in the dual effects of GLP-1 in alpha and beta cells. Our findings highlight unique contributions of individual Rb family members to islet cell proliferation and survival.
Similar content being viewed by others

Introduction
Glucose homeostasis is tightly regulated by insulin and glucagon secreted by pancreatic beta and alpha cells, respectively [1–3]. Accordingly, a proper ratio of beta and alpha cells is crucial to meet the challenges of metabolic stress. Reduced number and function of beta cells with alpha cell excess is a pathological hallmark in diabetes [4–6]. Beta cell regeneration, in principle, could be achieved by neogenesis of islet stem/progenitor cells [7] or self-replication of pre-existing beta cells [8]. However, islet progenitors are rare and virtually absent after birth, when most mature islet cells are post-mitotic [8, 9]. Therefore, increasing research has focused on boosting beta cell mass while ideally suppressing alpha cell hyperplasia. Understanding the mechanisms that control alpha and beta cell proliferation may lead to the development of novel diabetes therapies.
Cell proliferation is a finely tuned process that transits cells through specific restriction points [10]. Adult pancreatic islet cells are mostly in the quiescent/G1/0 state but can re-enter cell cycle following appropriate stimuli. For example, the glucagon-like peptide-1 (GLP-1) analogue, exendin-4, stimulates beta cell proliferation [11, 12] while preventing alpha cell expansion [13, 14]. However, the mechanisms by which GLP-1 mediates these divergent effects in alpha and beta cells are unknown.
Retinoblastoma protein (Rb) is a well-known tumour-suppressor and gatekeeper of cell cycle re-entry through inhibition of E2f transactivation and E2f target genes [10]. Rb, therefore, is a viable target for controlling islet cell cycle. Previous studies showed that Rb plays a critical role during the transition from proliferative to differentiated states, but has a limited role once cells exit the cell cycle and differentiate [15, 16]. Thus, Rb plays a reduced essential role in mature pancreatic beta cells [17], whereas disruption of Rb in proliferating progenitors improves glucose tolerance through its divergent role in alpha and beta cells [18].
Here we showed the essential role of Rb in the effects of GLP-1 in alpha and beta cells. We also observed reduced Rb in islets of humans with diabetes. To better understand the physiological role of Rb family proteins we assessed Rb homologue p107. Unlike Rb, p107 did not have essential roles in GLP-1 action or in alpha or beta cell homeostasis. However, p107 potentiated the effects of Rb such that combined deficiency increased apoptosis in both alpha and beta cells, and decreased islet mass with ageing. Our results show that Rb is essential in mediating the divergent actions of GLP-1 on alpha and beta cells, and together with its homologue p107, is critical in governing islet cell mass.
Methods
Mice
Pancreas-specific Rb knockout mice, Pdx1-Cre:Rb fl/fl (p-RbKO) [18], were bred to p107 −/− whole-body knockout mice (p107KO) (E. Zacksenhaus, Toronto, ON, Canada) [19] to generate Pdx1-Cre:Rb fl/fl/p107 −/− mice (p-DKO) and control Pdx1-Cre:Rb +/+/p107 +/+ littermates. Mice were maintained on a mixed C57BL6;129/Sv background and housed as previously described [18]. Exendin-4 (Sigma, St Louis, MO, USA) in PBS was administrated i.p. at 09:00 hours and 17:00 hours for 3 days at a dose of 24 nmol/kg [20]. Glucose tolerance tests (GTT), insulin tolerance tests (ITT) and glucose-stimulated insulin secretion (GSIS) were performed as previously described [21]. Animal protocols were approved by the Toronto General Research Institute Animal Care Committee.
Immunohistochemistry, immunofluorescence and immunoblotting
Ki67, insulin and glucagon immunostaining, TUNEL assay and immunoblotting were performed as described previously [22]. Antibodies for Rb, p107, E2F1, p53, p21, p27 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), cyclin D1, cyclin E, cleaved caspase 3, glyceraldehyde-3-phosphate dehydrogenase, p-Akt, and proliferating cell nuclear antigen (PCNA; Cell Signaling, Beverly, MA, USA) were used [22].
Cell culture, siRNA transfection, exendin-4 treatment and adenovirus infection
Insulinoma cells (INS-1) and simian virus 40 T-antigen induced glucagonoma cells (alpha-TC) were cultured as previously described [18]. Cells were serum-starved for 2 h and then treated with 10 nmol/l exendin-4. Cells were transfected with Rb, p107 or Silencer Select negative control small interfering RNA (siRNA) (Ambion, Carlsbad, CA, USA) [18]. Cells were infected with recombinant adenovirus-CMV-Rb (Vector Biolabs, Philadelphia, PA) at a multiplicity of infection of 100 for 24 h. Flow cytometry was performed as described previously [18].
Quantitative PCR
Human islets were provided by the ABCC Human islet distribution programme (University of Alberta, Edmonton, AB, Canada) [23, 24]. Islet donation was approved by the local institutional ethical review board. Quantitative PCR was performed as previously described [18].
Statistics
Data are presented as mean ± SEM and were analysed by two-tailed independent-sample Student’s t test or one-way ANOVA, as appropriate. A p value <0.05 was considered statistically significant.
Results
Critical role of Rb in mediating exendin-4 action in alpha and beta cells
Activating GLP-1 signalling is a widely used therapeutic strategy for diabetes that promotes beta cell proliferation while attenuating alpha cell mass in animal models [13]. We asked whether the opposing effects of GLP-1 on alpha and beta cells were mediated by Rb. We examined the direct effects of exendin-4 on beta cell (INS-1) and alpha cell (alpha-TC) lines. Exendin-4 induced an expected increase in cyclic AMP response element-binding protein phosphorylation, confirming activation of GLP-1 signalling in these cells (electronic supplementary material [ESM] Fig. 1a, b). In both cell lines exendin-4 decreased Rb protein levels and induced E2F1 (Fig. 1a, b). Phosphorylation of Rb was also induced as previously reported (ESM Fig. 1a, b) [12]. Rb levels did not change in response to high glucose, supporting its response specifically to exendin-4. Similar exendin-4-stimulated decrease in Rb was seen in isolated mouse islets (ESM Fig. 1c, d).

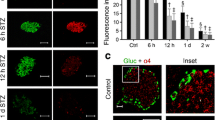
Dichotomous role of Rb in islet cycle. Western blots with quantification presented as fold vs control in (a) alpha-TC and (b) INS-1 cells after exendin-4 (EX); n = 3. (c) Arf mRNA levels fold vs control; n = 3. White bars, 0 h; grey bars, 6 h; black bars, 24 h. mRNA expression fold vs control in (d) Rb adenovirus (Ad-Rb)-infected alpha-TC and (e) INS-1 cells with 24 h EX; n = 3. White bars, control; grey bars, Ad-Rb; black bars, Ad-Rb + EX. (f) Ki67 positivity in 4–8-week-old mice; n = ~1,000 islet cells per animal, 3–5 mice. White bars, control; dark grey bars, RbKO; light grey bars, p107KO; black bars, p-DKO. *p < 0.05, **p < 0.01, ***p < 0.001 vs control in same treatment group by Student’s t test; ††† p < 0.001 vs same genotype group without EX by Student’s t test. (g) Human islet RB mRNA levels presented as fold vs control; n = 8 non-diabetic group (white bars) and n = 3 type 2 diabetic group (black bars). ***p < 0.001 between groups by Student’s t test. CCND1, cyclin D1; CCNE, Cyclin E; GAPDH, glyceraldehyde-3-phosphate dehydrogenase
Remarkably, the levels of E2F1 effectors in these two cell types were significantly different. In alpha-TC cells, cell cycle inhibitors, p53, p27 and p21 were increased, and cell cycle promoters cyclin D1 and E decreased (Fig. 1a); whereas in INS-1 cells, cell cycle inhibitors decreased with an increase in cyclin D1 and E (Fig. 1b). Accordingly, exendin-4 decreased viability in alpha-TC but increased viability in INS-1 cells (ESM Fig. 1e). To further understand the mechanism of these divergent effects of Rb, we next examined the expression of Arf, the E2F1 target and upstream regulator of p53. Interestingly, Arf was induced by exendin-4 in both cell types after 6 h but this induction was transient in beta cells. In alpha cells, Arf remained elevated at 24 h, potentially contributing to opposing p53 levels in the two cell types (Fig. 1c).
In order to assess whether or not Rb suppression was the central mechanism through which exendin-4 exerted its effects, we overexpressed Rb in both cell lines. Indeed, the effects on E2F1, cyclin D1 and E observed in both cells lines in response to exendin-4 were abolished with sustained Rb expression. In INS-1 cells with Rb overexpression, decreases in p53 and p21 were abrogated, and in alpha-TC cells with Rb overexpression, induction of cell cycle inhibitors by exendin was significantly attenuated. Attenuation of Arf induction in response to exendin-4 was also seen after Rb overexpression, supporting the important role of ADP-ribosylation factor (ARF) in mediating effects of Rb regulation. Overall, these data indicate the essential role of Rb suppression in the opposing biological effects of exendin-4 in these cell types (Fig. 1d, e).
To determine whether or not Rb is essential for GLP-1 effects in vivo, we next administered exendin-4 to mice with Rb deficiency specifically in the pancreas. We recently showed that Pdx1-Cre + Rb fl/fl mice (henceforth referred to as p-RbKO) with Rb deletion in pancreatic progenitors exhibit increased beta cell proliferation under basal conditions (Fig. 1f) [18]. As expected, exendin-4 treatment in control mice led to beta cell proliferation (Fig. 1f). By contrast, further induction of proliferation was not seen in p-RbKO mice (Fig. 1f), supporting the importance of Rb in GLP-1 action in vivo.
We next assessed the role of Rb homologue, p107, on exendin-4 response using whole-body p107KO mice (ESM Fig. 2a) given exendin-4. In contrast to p-RbKO, p107KO mice showed an increase in proliferation in response to exendin-4 similar to control mice (Fig. 1f). Furthermore, islets of mice with double deletion of Rb and p107 (p-DKO) showed similar lack of additional proliferative response to exendin-4, indicating that Rb but not p107 suppression was required for exendin-4 effects on islet cell proliferation (Fig. 1f).
Reduced Rb but not p107 expression in islets of humans with diabetes
Individuals with diabetes undergo adaptive islet cell proliferation to overcome insulin resistance [25]. Accordingly, a recent report showed upregulation of cell cycle genes CCND1 (encoding cyclin D1) and CDK4 (encoding cyclin-dependent kinase 4) in islets of humans with type 2 diabetes [26]. We, therefore, investigated whether Rb and p107 levels were altered with diabetes and found reduced RB expression in islets from individuals with diabetes compared with controls (Fig. 1g). Reduction of p107 was not statistically significant (Fig. 1g). Thus, Rb but not p107 is likely to be a critical negative regulator of adaptive beta cell proliferation.
Unique role of p107 in potentiating apoptosis in Rb-deficient islets
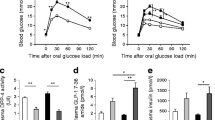
To better understand the role of Rb and p107 in islets, we assessed whether these proteins are essential for glucose homeostasis under basal conditions. We previously showed that p-RbKO mice have improved glucose tolerance and increased beta cell mass [18]. By contrast, p107KO mice had normal glucose tolerance, similar to controls, and p-DKO mice showed improved glucose tolerance similar to p-RbKO mice at 4–8 weeks of age (Fig. 2a). Interestingly, despite improved glucose tolerance, p-DKO mice did not have increased beta cell mass present in p-RbKO mice (Fig. 2b). While proliferation was similar to p-RbKO mice (Fig. 1f), there was a concomitant increase in beta cell apoptosis in p-DKO mice, as shown by TUNEL and cleaved caspase 3 in islet lysates (Fig. 2c, d). Therefore, increased apoptosis may explain the lack of increase in beta cell mass in p-DKO mice (Fig. 2b). Interestingly, beta cell function, as evidenced by GSIS, was enhanced in p-DKO mice (Fig. 2e), and significantly more so than in p-RbKO mice, which were previously shown to have increased beta cell function. This improved glucose tolerance in p-DKO mice was not due to changes in insulin sensitivity as we observed similar body weights and response to ITT compared with controls (ESM Fig. 2b, c). Therefore, the enhanced beta cell function in p-DKO mice likely contributed to the improved glucose tolerance despite lower beta cell mass.

Rb/p107 deficiency in islets improves glucose tolerance in young (4–8-week-old) mice. (a) GTT; n = 8. (b) Beta cell area; n = 5. (c) Pancreas TUNEL/insulin co-staining. Arrowheads, TUNEL-positive beta cells; n = ~1000 beta cells per animal, 5 mice. (d) Protein levels presented as fold vs control of cleaved caspase 3 in islets; n = 3. (e) GSIS; n = 8. White circles, control; white diamonds, RbKO; grey circles, p107KO; black circles, p-DKO. White bars, control; dark grey bars, RbKO; light grey bars, p107KO; black bars, p-DKO. *p < 0.05, **p < 0.01, ***p < 0.001 p-RbKO vs controls by Student’s t test; † p < 0.05, †† p < 0.01 p-DKO vs controls by Student’s t test. Scale bar, 40 μm
Interestingly, as p-DKO mice aged they no longer exhibited enhanced glucose tolerance, whereas p-RbKO mice maintained better glycaemic control (18–22 weeks; Fig. 3a, b). This loss of improved glucose homeostasis in older p-DKO mice correlated with reduced beta cell mass (Fig. 3c) and increased beta cell apoptosis, similar to that seen in younger mice (4–8 weeks). However, the increased proliferation present in younger p-DKO mice was absent in older mice. Thus the combined increase in apoptosis and a decline in beta cell proliferation likely contributed to the overall decrease in beta cell mass in these mice with ageing and decreased insulin levels during GSIS (Fig. 3d–f, ESM Fig. 2d). On the other hand, p107KO mice had no change in beta cell proliferation or apoptosis at both ages (Figs 1f, 2d, and 3d), suggesting that p107 is not essential in beta cell homeostasis. However, p107 potentiates apoptosis in beta cells when combined with Rb deficiency.

Loss of beta cell mass in aged (18–22 weeks) Rb/p107-deficient mice. (a) Blood glucose levels; n = 10. (b) GTT; n = 9. (c) Beta cell area; n = 5. (d) Islet Ki67 positivity; n = 5. (e) Pancreas TUNEL/insulin co-staining. Arrowheads, TUNEL-positive beta cells; n = ~1000 beta cells per animal, 5 mice. (f) GSIS; n = 5. White circles, control; white diamonds, RbKO; grey circles, p107KO; black circles, p-DKO. White bars, control; dark grey bars, RbKO; light grey bars, p107KO; black bars, p-DKO. *p < 0.05, ***p < 0.001 vs control by Student’s t test; † p < 0.05, †† p < 0.01 p-RbKO vs controls by Student’s t test. Scale bar, 40 μm
We next assessed the role of p107 in alpha cells. We previously showed that Rb deficiency in islet progenitors lowered alpha cell mass. In p-DKO mice, there was a significant further reduction in alpha cell mass in both age groups compared with p-RbKO mice (Fig. 4a, b), which was associated with increased apoptosis, evident from TUNEL of glucagon-expressing cells (Fig. 4c). We previously showed that Rb controls alpha cell development through regulation of the key transcription factor Arx. While Arx was not changed in p107 deficient islets, its expression was further reduced in p-DKO compared with Rb-deficient islets (Fig. 4d). These changes in alpha cells were in line with a significant reduction in fasting serum glucagon levels in p-DKO mice (Fig. 4e). The reduction in alpha cells could also contribute to improved glucose tolerance in the young mice, as well as to sustained normal glucose tolerance despite low beta cell mass in the older p-DKO mice (Figs 2a and 3b). Therefore, while loss of p107 alone does not alter islet homeostasis, likely due to compensation by Rb, when combined with Rb deletion, it renders both alpha and beta cells more susceptible to apoptosis, ultimately resulting in a decline in islet mass and glucose intolerance.

Loss of alpha cell mass in Rb/p107-deficient mice. (a) Pancreas insulin/glucagon co-staining; n = 3. (b) alpha cell area; n = 5. (c) Pancreas TUNEL/glucagon co-staining. Arrowheads, TUNEL-positive alpha cells; n = ~100 alpha cells per animal, 3–5 mice. (d) Islet Arx mRNA levels presented as fold vs control; n = 3. (e) Fasting serum glucagon; n = 5. White bars, control; dark grey bars, RbKO; light grey bars, p107KO; black bars, p-DKO. *p < 0.05, **p < 0.01, ***p < 0.001 vs control by Student’s t test; † p < 0.05, †† p < 0.01 p-DKO vs p-RbKO mice by Student’s t test. Scale bar, 40 μm
Disruption of Rb and p107 in alpha and beta cells leads to deregulation of E2F
E2F members have a dose-dependent effect on cell fate, leading to either proliferation or apoptosis [27–30]. Indeed, at 4–8 weeks of age, p-DKO islets expressed much higher levels of E2f1, 4 and 5 compared with their single knockout counterparts (Fig. 5a). This finding was associated not only with an increase in pro-survival and proliferative signalling, p-Akt, cyclin E and PCNA, but also with a concomitant increase in cell cycle inhibitors p53, p27 and p21 (Fig. 5b), which was not seen in the p-RbKO mice (ESM Fig. 2e) [18]. Furthermore, E2F1 is required for the divergent effects of exendin-4 on alpha and beta cell lines, since siRNA knockdown of E2F1 abolished the changes in expression of cell cycle regulatory genes seen with exendin-4 (ESM Fig. 3a, b).

Altered levels of cell cycle regulators in p-DKO mice. (a, c) Islet mRNA expression presented as fold vs control; n = 3. (b) Islet protein levels presented as fold vs control; n = 3. White bars, control; dark grey bars, RbKO; light grey bars, p107KO; black bars, p-DKO. *p < 0.05, **p < 0.01, ***p < 0.001 vs control by Student’s t test; †† p < 0.01 vs RbKO by Student's t test; ‡ p < 0.05, ‡‡ p < 0.01 vs p107KO by Student’s t test. CCNE, cyclin E
The increased p53 levels seen in islets from the p-DKO mice but not from single knockout mice were associated with induction of its pro-death targets, Puma (also known as Bbc3) and Noxa (also known as Pmaip1) (Fig. 5c). Moreover, in contrast to the single knockout mice, p-DKO mouse islets had decreased expression of the anti-apoptotic Bcl-X L (also known as Bcl2l1) gene as well as a decrease in P16ink4a (also known as Cdkn2a), involved in beta cell regeneration [31]. These findings suggest that combined Rb and p107 deficiency further alters cell survival and proliferation. Overall, both alpha and beta cell apoptosis were increased (Figs 2c and 3e), in addition to an increase in proliferation in p-DKO islets, resulting in similar islet mass as controls. By contrast, p-RbKO islets with lower levels of E2Fs compared with p-DKO islets showed reduced Puma and Noxa mRNA levels, and an increase in Bcl-X L mRNA, which encodes an anti-apoptotic protein (Fig. 5c).
We next assessed the cell-autonomous role of p107 either alone or in combination with Rb using siRNA in alpha and beta cell lines. Consistent with the in vivo data, knockdown of p107 in alpha-TC cells did not induce p53 (Fig. 6a). However, when p107 and Rb were both knocked down, p53 was increased (Fig. 6a), associated with even higher apoptotic SubG1 fraction compared with Rb knockdown alone (Fig. 6b).

Knockdown of Rb and p107 increases apoptosis. Protein levels presented as fold vs control in (a) alpha-TC and (c) INS-1 cells; n = 3. Cell cycle analyses of (b) alpha-TC and (d) INS-1 cells by flow cytometry; n = 3. (e) Arf and E2f1 mRNA levels presented as fold vs control in siRNA-treated cells; n = 3. White bars, control; dark grey bars, scramble siRNA; striped bars, Rb siRNA; dark grey bars, p107 siRNA; black bars, Rb/p107 siRNA. (f) Islet Arf mRNA level; n = 3. White bars, control; dark grey bars, RbKO; light grey bars, p107KO; black bars, p-DKO. *p < 0.05, **p < 0.01 vs control by Student’s t test; † p < 0.05 vs p107KO by Student’s t test. FL2-A, area; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PI, propidium iodide
In INS-1 cells, combined knockdown of Rb and p107 induced p53 to a much greater extent than with single-gene knockdown (Fig. 6c), substantially increasing apoptotic SubG1 fraction (Fig. 6d), while knockdown of Rb alone led to p53 suppression without significant changes in apoptosis (Fig. 6d). Interestingly, Arf expression increased in both alpha and beta cells after combined Rb and p107 knockdown (Fig. 6e). Consistent with this, we observed increased Arf expression and apoptosis in islets of p-DKO mice (Fig. 6f). We also observed increases in Kir6.2 (also known as Kcnj11) expression in p-Rb and p-DKO mice, which is well known for regulation in insulin secretion and has also been implicated in beta cell proliferation, consistent with its regulation by E2F1 (ESM Fig. 3c) [32, 33]. These results show intricate regulation of p53 by Rb family proteins and the fine-tuning of intracellular signalling that is potentiated by Rb and p107.
Discussion
Dysregulation of pancreatic islet cell mass is at the crux of diabetes mellitus. Thus regenerating islets with proper proportions of individual islet cell types would be an ideal goal for successful treatment of diabetes. In this study, we observed that Rb levels were decreased in islets of humans with type 2 diabetes, suggesting that Rb is important in compensatory beta cell proliferation. Interestingly, exendin-4 treatment of beta cells also suppressed Rb, an effect necessary for the proliferative effects of this agent. Exendin-4 treatment of alpha cells similarly reduced Rb levels but this led to cell cycle arrest, which may have further contributed to metabolically favourable effects of exendin-4. Exendin-4 suppressed Rb through two complementary mechanisms: downregulation of Rb mRNA and increased Rb phosphorylation. While Rb hyperphosphorylation may be driven through exendin 4-dependent induction of cyclin D1 [34], the mechanism by which this GLP-1R agonist inhibits Rb mRNA is yet to be established. The predominant Rb family member expressed in cycling cells, p107, showed a limited role in adult beta cell cycle control. However, combined p107 and Rb deficiency in islet progenitors drove beta cells from a quiescent state back into cell cycle; but this effect was overcome by an increase in apoptosis with ageing. These results show intricate roles of the individual and combined Rb family members in regulating islet cell cycle and survival (Fig. 7).

Summary of the role of Rb and p107 in alpha (α) and beta (β) cell cycle control
Rb is thought to be dispensable in mature beta cells, possibly due to compensation by p130 [17, 35]. However, deletion of Rb in proliferating pancreatic and duodenal homeobox 1 (PDX1)-positive pancreatic progenitors improved glucose tolerance by persistently increasing neurogenin 3-positive cells that were associated with enhanced beta cell proliferation and differentiation, but disrupted alpha cell differentiation and survival [18]. We show that combined deletion of Rb and p107 in proliferating islets does not exert additional proliferative effects compared with loss of Rb alone in beta cells; however, ageing increases beta cell apoptosis, leading to loss of beta cell mass and abolition of enhanced glucose homeostasis. These results demonstrate a unique role of p107 deletion in potentiating islet cell apoptosis in Rb-deficient islets through induction of E2F members and p53, thereby resulting in a decline of both alpha and beta cell mass.
Rb and p107 may compensate for each other through two major mechanisms. First, in the absence of Rb, p107 may interact with activating E2Fs (E2F1–3) and suppress genes normally regulated by Rb. Thus, combined inactivation of both pocket proteins is required to deregulate E2F1–3-responsive genes and promote cell cycle progression. Second, the spacer domain of p107 (but not Rb) acts as a CDK2 inhibitor [36]. Indeed, deletion of p107 has a similar effect as deletion of the CDK inhibitor p27 on retinoblastoma formation in murine models [37]. Deletion of Rb and p107 in some contexts leads to enhanced proliferation and cancer such as retinoblastoma in mice, but may also lead to more severe differentiation defects in muscle [38] or, as shown here, to increased apoptosis in pancreatic islet cells.
The distinct biological consequences of the loss of specific Rb family members can be due to differential regulation of E2F family proteins [10, 39]. For example, in alpha cells, Rb knockdown led to an approximately fivefold increase in E2F1 levels, which was associated with p53 induction and an increase in apoptosis. By contrast, a less dramatic twofold induction of E2F1 after a similarly efficient knockdown of Rb in beta cells resulted in a decrease in p53 with an increase in cell proliferation [18]. Similarly, a mild elevation of E2F1 levels in fibroblasts resulted in increased cell proliferation, but an excessive increase in E2F1 led to apoptosis through activation of p53-dependent caspase signalling [27]. These findings suggest that E2F1 may have a dose-dependent effect on cell fate. Accordingly, we show that combined deletion of Rb and p107 in beta cells led to a greater increase in E2F1 expression compared with loss of Rb alone, which not only increased proliferation but also apoptosis. Indeed, E2F1 is considered unique from other E2F members because of its effect in promoting pro-apoptotic activity [29, 30]. Persistently increased apoptosis with a concomitant reduction in proliferation resulted in net loss of beta cell mass in p-DKO mice with ageing, leading to the loss of improved glucose homeostasis observed in young p-DKO mice. Moreover, E2F1 has previously been shown to be involved in early pancreas development [40], insulin secretion [33, 41] and proliferation [41–44]. These data show the importance of the precise regulation of E2F in determining the various biological outcomes of islets.
The differential induction of E2F1 can in turn lead to specific regulation of Arf, which could contribute to divergent cell fate. Arf was transiently induced in exendin-4-treated beta cells, returning to baseline levels after 24 h, whereas high Arf levels persisted in alpha cells following treatment, and these inductions were attenuated with Rb overexpression. These results show a distinct dynamic response of Arf in alpha and beta cells, which may contribute to dose-dependent effects of E2F1 on islet cell fate. While E2F1 is perhaps best recognised as a major target of Rb regulation in cell cycle, further work is required to determine the potential roles of other E2Fs, which have increasingly been shown to play a role in cell survival and cycle regulation [45], including E2F3a and E2F3b which have been implicated in cell survival and Arf regulation [46, 47].
In addition, we found that combined loss of Rb and p107 in alpha cells led to an even greater increase in apoptosis, as evidenced by an approximately twofold increase in alpha cell apoptosis in p-DKO vs p-RbKO mice. We showed previously that E2F1 can bind directly to the promoter regions of Arx, a key alpha cell transcription factor, and likely represses gene transcription. As such, Rb-deficient mice exhibit low alpha cell mass [18]. The more significant loss in alpha cell mass in p-DKO mice suggests that the combined loss of Rb family proteins potentiates Arx repression and alpha cell death. The profound reduction in alpha cell mass in p-DKO mice likely contributed to improved glucose homeostasis in young mice with normal beta cell mass. Moreover, the reduced alpha cell mass is also likely to have contributed to normal glucose tolerance in aged p-DKO mice with decreased beta cell mass.
As the gatekeeper of cell cycle entry, Rb proteins play a critical role in determining the transition between proliferation, differentiation and apoptosis [16, 48]. A recent report has demonstrated that another typically non-proliferative and post-mitotic cell type, muscle cells, can become regenerative by inactivating Rb and ARF [49]. Moreover, expansion of stem cell populations have also been shown to be restricted by the Rb pathway [50]. Combined with our results in human and mouse islets, these lines of evidence further support the notion that differentiated mammalian cells still retain the capacity to re-enter cell cycle and this regenerative ability may be largely controlled by the activities of Rb proteins.
The results of the present study demonstrate that Rb family proteins have unique and opposing effects on alpha and beta cells that provide a unifying mechanism for the divergent role of GLP-1 in these islet cell types. In contrast to the opposing effects of Rb loss alone, the Rb homologue p107, while not essential by itself, augments apoptosis in both alpha and beta cells in the setting of combined Rb and p107 loss. Given that a decline in beta cells and concomitant increase in alpha cells are core pathologies in diabetes, our findings on the dichotomous effects of Rb in alpha and beta cells provide valuable insight towards novel therapeutic strategies.
Abbreviations
- ARF:
-
ADP-ribosylation factor
- CDK:
-
Cyclin-dependent kinase
- GLP-1:
-
Glucagon-like peptide-1
- GSIS:
-
Glucose-stimulated insulin secretion
- GTT:
-
Glucose tolerance test
- ITT:
-
Insulin tolerance test
- PCNA:
-
Proliferating cell nuclear antigen
- p-DKO:
-
Rb/p107 double-knockout
- p107KO:
-
Whole-body p107 knockout
- p-RbKO:
-
Pancreas-specific Rb knockout
- Rb:
-
Retinoblastoma protein
- siRNA:
-
Small interfering RNA
References
Meier JJ, Kjems LL, Veldhuis JD, Lefebvre P, Butler PC (2006) Postprandial suppression of glucagon secretion depends on intact pulsatile insulin secretion: further evidence for the intraislet insulin hypothesis. Diabetes 55:1051–1056
Quesada I, Tuduri E, Ripoll C, Nadal A (2008) Physiology of the pancreatic alpha-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol 199:5–19
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806
Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL (2005) Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54(Suppl 2):S97–S107
Unger RH, Aguilar-Parada E, Muller WA, Eisentraut AM (1970) Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Invest 49:837–848
Unger RH, Orci L (1977) The role of glucagon in the endogenous hyperglycemia of diabetes mellitus. Annu Rev Med 28:119–130
Xu X, D'Hoker J, Stange G et al (2008) Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132:197–207
Dor Y, Brown J, Martinez OI, Melton DA (2004) Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429:41–46
Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA (2005) Very slow turnover of beta-cells in aged adult mice. Diabetes 54:2557–2567
Knudsen ES, Knudsen KE (2008) Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 8:714–724
Song WJ, Schreiber WE, Zhong E et al (2008) Exendin-4 stimulation of cyclin A2 in beta-cell proliferation. Diabetes 57:2371–2381
Kim MJ, Kang JH, Park YG et al (2006) Exendin-4 induction of cyclin D1 expression in INS-1 beta-cells: involvement of cAMP-responsive element. J Endocrinol 188:623–633
Takeda Y, Fujita Y, Honjo J et al (2012) Reduction of both beta cell death and alpha cell proliferation by dipeptidyl peptidase-4 inhibition in a streptozotocin-induced model of diabetes in mice. Diabetologia 55:404–412
Ellenbroek JH, Tons HA, Westerouen van Meeteren MJ et al (2013) Glucagon-like peptide-1 receptor agonist treatment reduces beta cell mass in normoglycaemic mice. Diabetologia 56:1980–1986
Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL (1996) pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev 10:3051–3064
Huh MS, Parker MH, Scime A, Parks R, Rudnicki MA (2004) Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J Cell Biol 166:865–876
Vasavada RC, Cozar-Castellano I, Sipula D, Stewart AF (2007) Tissue-specific deletion of the retinoblastoma protein in the pancreatic beta-cell has limited effects on beta-cell replication, mass, and function. Diabetes 56:57–64
Cai EP, Wu X, Schroer SA et al (2013) Retinoblastoma tumor suppressor protein in pancreatic progenitors controls alpha- and beta-cell fate. Proc Natl Acad Sci U S A 110:14723–14728
Jiang Z, Zacksenhaus E, Gallie BL, Phillips RA (1997) The retinoblastoma gene family is differentially expressed during embryogenesis. Oncogene 14:1789–1797
Choi D, Radziszewska A, Schroer SA et al (2009) Deletion of Fas in the pancreatic beta-cells leads to enhanced insulin secretion. Am J Physiol Endocrinol Metab 297:E1304–E1312
Choi D, Schroer SA, Lu SY et al (2010) Erythropoietin protects against diabetes through direct effects on pancreatic beta cells. J Exp Med 207:2831–2842
Cai EP, Casimir M, Schroer SA et al (2012) In vivo role of focal adhesion kinase in regulating pancreatic beta-cell mass and function through insulin signaling, actin dynamics, and granule trafficking. Diabetes 61:1708–1718
Shapiro AM, Lakey JR, Ryan EA et al (2000) Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 343:230–238
Basford CL, Prentice KJ, Hardy AB et al (2012) The functional and molecular characterisation of human embryonic stem cell-derived insulin-positive cells compared with adult pancreatic beta cells. Diabetologia 55:358–371
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444:840–846
Taneera J, Fadista J, Ahlqvist E et al (2013) Expression profiling of cell cycle genes in human pancreatic islets with and without type 2 diabetes. Mol Cell Endocrinol 375:35–42
Nahle Z, Polakoff J, Davuluri RV et al (2002) Direct coupling of the cell cycle and cell death machinery by E2F. Nat Cell Biol 4:859–864
Qin XQ, Livingston DM, Kaelin WG Jr, Adams PD (1994) Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci U S A 91:10918–10922
DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR (1997) Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci U S A 94:7245–7250
Rogoff HA, Pickering MT, Frame FM et al (2004) Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol 24:2968–2977
Krishnamurthy J, Ramsey MR, Ligon KL et al (2006) p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443:453–457
Porat S, Weinberg-Corem N, Tornovsky-Babaey S et al (2011) Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab 13:440–449
Annicotte JS, Blanchet E, Chavey C et al (2009) The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat Cell Biol 11:1017–1023
Kang JH, Kim MJ, Ko SH et al (2006) Upregulation of rat Ccnd1 gene by exendin-4 in pancreatic beta cell line INS-1: interaction of early growth response-1 with cis-regulatory element. Diabetologia 49:969–979
Harb G, Vasavada RC, Cobrinik D, Stewart AF (2009) The retinoblastoma protein and its homolog p130 regulate the G1/S transition in pancreatic beta-cells. Diabetes 58:1852–1862
Cobrinik D (2005) Pocket proteins and cell cycle control. Oncogene 24:2796–2809
Sangwan M, McCurdy SR, Livne-Bar I et al (2012) Established and new mouse models reveal E2f1 and Cdk2 dependency of retinoblastoma, and expose effective strategies to block tumor initiation. Oncogene 31:5019–5028
Ciavarra G, Ho AT, Cobrinik D, Zacksenhaus E (2011) Critical role of the Rb family in myoblast survival and fusion. PLoS One 6:e17682
Iaquinta PJ, Lees JA (2007) Life and death decisions by the E2F transcription factors. Curr Opin Cell Biol 19:649–657
Kim SY, Rane SG (2011) The Cdk4-E2f1 pathway regulates early pancreas development by targeting Pdx1+ progenitors and Ngn3+ endocrine precursors. Development 138:1903–1912
Fajas L, Annicotte JS, Miard S, Sarruf D, Watanabe M, Auwerx J (2004) Impaired pancreatic growth, beta cell mass, and beta cell function in E2F1 (-/-) mice. J Clin Invest 113:1288–1295
Li FX, Zhu JW, Tessem JS et al (2003) The development of diabetes in E2f1/E2f2 mutant mice reveals important roles for bone marrow-derived cells in preventing islet cell loss. Proc Natl Acad Sci U S A 100:12935–12940
Iglesias A, Murga M, Laresgoiti U et al (2004) Diabetes and exocrine pancreatic insufficiency in E2F1/E2F2 double-mutant mice. J Clin Invest 113:1398–1407
Grouwels G, Cai Y, Hoebeke I et al (2010) Ectopic expression of E2F1 stimulates beta-cell proliferation and function. Diabetes 59:1435–1444
Dick FA, Rubin SM (2013) Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol 14:297–306
Danielian PS, Friesenhahn LB, Faust AM et al (2008) E2f3a and E2f3b make overlapping but different contributions to total E2f3 activity. Oncogene 27:6561–6570
Miki J, Fujimura Y, Koseki H, Kamijo T (2007) Polycomb complexes regulate cellular senescence by repression of ARF in cooperation with E2F3. Genes Cells 12:1371–1382
Ikeda MA, Jakoi L, Nevins JR (1996) A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc Natl Acad Sci U S A 93:3215–3220
Pajcini KV, Corbel SY, Sage J, Pomerantz JH, Blau HM (2010) Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell Stem Cell 7:198–213
Sage J (2012) The retinoblastoma tumor suppressor and stem cell biology. Genes Dev 26:1409–1420
Acknowledgements
We thank T. Jin (Department of Physiology, University of Toronto, Toronto, ON, Canada) for providing INS-1 and alpha-TC cell lines. We thank M. Wheeler (Department of Physiology, University of Toronto, Toronto, ON, Canada) for providing human islet cDNA and K. J. Prentice for human islet cDNA preparation.
Funding
This work was supported by Canadian Institute of Health Research (CIHR) operating grants MOP-81148 and CCI-125690 to MW. EPC is supported by the Canadian Diabetes Association (CDA) Doctoral Student Research Award. CTL is supported by the Eliot Phillipson Clinician Scientist Training Program and postdoctoral fellowships from the Banting and Best Diabetes Centre (BBDC) and the CDA. SYS is supported by the Canadian Liver Foundation Graduate Studentship, CDA Doctoral Student Research Award and CIHR–Frederick Banting and Charles Best Canada Graduate Scholarship. TS is supported by a BBDC Novo Nordisk Studentship, CDA Doctoral Student Research Award and CIHR – Frederick Banting and Charles Best Canada Graduate Scholarship. JJB was supported by a BBDC Novo Nordisk Studentship. MW is supported by the Canada Research Chair in Signal Transduction in Diabetes Pathogenesis.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors contributed to the study conception and data, and approved the final version of the manuscript. EPC and CTL contributed to the generation and analyses of research data, and preparation of the manuscript. XW, SAS, SYS, TS and JJB contributed to the generation of research data and reviewed the manuscript. EZ and MW designed experiments, supervised students, contributed to discussion and interpretation of the data, and reviewed and edited the manuscript. MW is the guarantor of this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 3177 kb)
ESM Fig. 2
(PDF 4641 kb)
ESM Fig. 3
(PDF 560 kb)
Rights and permissions
About this article

Cite this article
Cai, E.P., Luk, C.T., Wu, X. et al. Rb and p107 are required for alpha cell survival, beta cell cycle control and glucagon-like peptide-1 action. Diabetologia 57, 2555–2565 (2014). https://doi.org/10.1007/s00125-014-3381-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3381-y