
Abstract
Aims/hypothesis
Glucagon-like peptide-1 receptor (GLP-1R) agonists improve glucose control in animals and humans with type 1 diabetes. However, there is little information on the role of the GLP-1R in the immune system. We studied the role of the GLP-1R in immune function in wild-type (WT) and non-obese diabetic (NOD) and Glp1r −/− mice.
Methods
Glp1r mRNA expression was examined in sorted immune subpopulations by RT–PCR. The effects of GLP-1R activation were assessed on cAMP production and proliferation, migration and survival of primary immune cells from WT and NOD mice. The ability of primary cells from Glp1r −/− mice to proliferate, migrate or survive apoptosis was determined. Immunophenotyping studies were performed to assess the frequency of immune subpopulations in Glp1r −/− mice.
Results
Ex vivo activation of the GLP-1R resulted in a modest but significant elevation of cAMP in primary thymocytes and splenocytes from both WT and NOD mice. GLP-1R activation did not increase proliferation of primary thymocytes, splenocytes or peripheral lymph node cells. In contrast, Glp1r −/− thymocytes exhibited a hypoproliferative response, whilst peripheral Glp1r −/− lymphocytes were hyperproliferative in response to mitogenic stimulation. Activation or loss of GLP-1R signalling did not modify apoptosis or chemotaxis in primary lymphocytes. Male Glp1r −/− mice exhibited a significantly lower percentage of peripheral regulatory T cells, although no differences were observed in the numbers of CD4+ and CD8+ T cells and B cells in the spleen and lymph nodes of Glp1r −/− mice.
Conclusions/interpretation
These studies establish that GLP-1R signalling may regulate lymphocyte proliferation and maintenance of peripheral regulatory T cells.
Similar content being viewed by others


Introduction
Glucagon-like peptide-1 receptor (GLP-1R) activation, either by preventing degradation of its endogenous ligand, or via administration of long-acting receptor agonists, is used for the treatment of type 2 diabetes [1]. GLP-1R activation induces multiple glucoregulatory actions, including inhibition of glucagon secretion and gastric emptying, both of which reduce postprandial glycaemic excursions in patients with type 1 diabetes [2]. The regenerative and cytoprotective effects of glucagon-like peptide-1 (GLP-1) on beta cells have engendered considerable interest in whether GLP-1 may be useful for the treatment of type 1 diabetes, either in combination with immunomodulation [3–6] or as adjunctive therapy in islet transplantation [7–10].
Intriguingly, GLP-1R agonists improve type 1 diabetes in animal models without the need for immunotherapy, thus implicating GLP-1R-dependent signalling pathways in immunoregulatory processes [11–14]. Nevertheless, little is known about the effects of GLP-1 on the immune system. Glp1r mRNA transcripts have been detected in murine lymphoid tissue [12], and GLP-1R activation stimulates regulatory T cells (Tregs), by both increasing the frequency and improving the function of these cells in recently diagnosed diabetic NOD mice [15].
The use of exenatide therapy in patients with type 1 diabetes [16, 17], despite limited knowledge of the putative immunoregulatory role of the GLP-1R, prompted us to ascertain whether the GLP-1R is active in a range of immune cells. We have now assessed immune function following GLP-1R activation in wild-type (WT) and non-obese diabetic (NOD) mice, a model of type 1 diabetes. In complementary studies, we characterised selective functional variables using primary immune cells from Glp1r −/− mice. Our data reveal a role for GLP-1R signalling in the proliferation and regulation of primary murine lymphocytes.
Methods
Reagents
Synthetic exendin-4 (Ex-4) was purchased from California Peptide Research (Napa, CA, USA), dissolved in phosphate buffered saline (pH 7.4) and stored at −80°C until use. GLP-1 (1–37), GLP-1 (7–36) amide and GLP-1 (9–36) were purchased from Bachem (Torrance, CA, USA). Prostaglandin E2 (PGE-2) was purchased from Sigma-Aldrich (St Louis, MO, USA).
Anti-CD3 and anti-CD28 antibodies were from BD Pharmingen (San Jose, CA, USA). Concanavalin A (ConA), phorbol myristate acetate (PMA) and ionomycin were from Sigma-Aldrich. IL-2, chemokine (C-C motif) ligand 19 (CCL19) (also known as macrophage inflammatory protein 3b [MIP-3b]), chemokine (C-X-C motif) ligand 12 (CXCL12) (also known as stromal cell-derived factor 1a [SDF1a]) and chemokine (C-C motif) ligand 25 (CCL25) were from Peprotech (Rocky Hill, NJ, USA). Dexamethasone was from Sigma-Aldrich, staurosporine from Calbiochem (Darmstadt, Germany) and anti-Fas antibody (Jo2) and annexin-V/7-amino-actinomycin D (7-AAD) apoptosis detection kit from BD Pharmingen.
For immunophenotyping and cell-sorting studies, the following monoclonal antibodies were prepared as described previously [18]: purified anti-CD16/CD32 (for Fc blocking), biotinylated anti-CD8α, FITC-labelled anti-CD8α, FITC-labelled anti-CD4, phycoerythrin (PE)-labelled anti-CD4, allophycocyanin (APC)-labelled anti-H57 (T cell receptor [TCR] β chain) and FITC-labelled anti-CD19. Streptavidin–Spectral Red (SPRD) was from Southern Biotech (Birmingham, AL, USA). FITC-labelled anti-IgM and PE-labelled anti-protein tyrosine phosphatase, receptor type, C (B220) were from BD Pharmingen. The mouse Treg-staining kit (FITC-labelled anti-CD4, PE-labelled anti-CD25 and APC-labelled anti-mouse/rat forkhead box protein [FOXP3] [FJK-16s]) was from eBioscience (San Diego, CA, USA).
Mice
NOD/Ltj mice were from the Jackson Laboratory (Bar Harbor, MI, USA) and C57Bl/6 mice were from Taconic Farms (Germantown, NY, USA). Mice were acclimatised to the animal facility for at least 1 week prior to analysis. For the immunophenotyping and loss-of-function studies, Glp1r −/− mice and age- and sex-matched littermate controls on a C57Bl/6 background were used. Mice were housed in a pathogen-free facility and maintained on a 12 h light–dark cycle, with free access to standard rodent chow and water. All experiments were carried out in accordance with protocols and guidelines approved by the Animal Care Committees of the Toronto General and Mt Sinai Hospitals.
FACS
Primary single-cell suspensions from thymus, bone marrow, lymph nodes and spleen of 6- to 9-week-old male and female NOD/Ltj and age-matched C57Bl/6 mice were prepared by passing the tissue through a 70 µm cell strainer and recovering cells in calcium- and magnesium-free Hanks’ balanced salt solution with 2% (vol./vol.) FBS and 10 mmol/l HEPES. For the bone marrow preparation, cells were flushed from the tibia and femur and clumps were dissociated by repeatedly passing the cells through a 10 ml syringe, then through a 70 µm cell strainer. Erythrocytes were lysed using RBC Lysis Buffer (Biolegend, San Diego, CA, USA) and removed from cell suspensions. Cell numbers were quantified using a haemocytometer. Cells were incubated with antibodies for 30 min at 4°C in staining media. After removing excess antibody, cells were filtered to remove clumps and sorted on a FACS Aria cell sorter (BD Biosciences, San Jose, CA, USA).
Glp1r expression in sorted immune subsets
Following cell sorting, cells were washed and lysed, and total RNA extracted using RNeasy Mini or Micro kits (Qiagen, Mississauga, ON, Canada). Target cDNA was analysed for the expression of mouse Glp1r and β-actin gene mRNA transcripts by the PCR method [12]. Primer pairs are indicated in Electronic supplementary material (ESM) Table 1. The products were visualised with SYBR Safe DNA stain (Invitrogen, Carlsbad, CA, USA) and transferred to a Nytran Super Charge Nylon Membrane (Mandel Scientific, Guelph, ON, Canada). The amplified cDNA was hybridised overnight using internal primers (ESM Table 1), as described previously [12].
Immunophenotyping analysis
Single-cell suspensions were prepared from the spleen, lymph nodes, thymus and bone marrow of Glp1r −/− and littermate control mice, as described above. Cells (106) were incubated at 4°C with primary and secondary (in the case of biotinylated primary) antibodies in cold staining media. Cells were either resuspended in cold staining media containing propidium iodide (for dead cell exclusion) and analysed directly, or fixed/permeabilised and stained with the anti-FOXP3 antibody according to the manufacturer’s protocol. Cells were acquired on a FACSCalibur flow cytometer (BD Biosciences) and analysed using FlowJo software (Tree Star, Ashland, OR, USA).
cAMP measurements
Single-cell suspensions from spleens, thymuses and lymph nodes of 10- to 12-week-old normoglycaemic female NOD/Ltj and C57Bl/6 mice were prepared as described above. Primary cells were resuspended in Earle’s balanced salt solution (Invitrogen) with 0.5% (vol./vol.) FBS and 10 mmol/l HEPES (treatment media). Triplicate wells were treated with the appropriate agonists, supplemented with 10 µmol/l 3-isobutyl-1-methylxanthine (Sigma-Aldrich), for 15 min in an incubator. The reaction was terminated by the addition of 65% (vol./vol.) cold ethanol. cAMP levels were measured with a cAMP RIA kit (Biomedical Technologies, Stoughton, MA, USA).
Proliferation studies
Primary thymocytes and lymph node cells were prepared aseptically in complete RPMI media with 10% (vol./vol.) heat-inactivated FBS, 10 mmol/l HEPES, 0.055 mmol/l 2-mercaptoethanol, 2 mmol/l L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. The indicated reagents were prepared in complete RPMI and added in triplicate in wells of tissue-culture (TC)-treated flat-bottom 96 well plates (BD Labware, Franklin Lakes, NJ, USA); 1 × 106 thymocytes and 5 × 105 lymph node cells were added per well and incubated for 48 h at 37°C in 5% CO2. Cells were then pulsed with 0.037 MBq [3H]thymidine per well (GE Healthcare, Piscataway, NJ, USA) and plates were returned to the incubator for an additional 18 h. Following cell harvesting, the incorporated radioactivity was measured using an automated β-scintillation counter. Data are expressed as a stimulation index, calculated as the mean cpm of treated wells divided by the mean cpm of negative control (untreated) wells.
Transwell migration assays
Assays were performed as previously described, with slight modifications [19]. Primary cells were recovered from spleens, thymuses and peripheral and mesenteric lymph nodes under sterile conditions, and resuspended in migration assay media (RPMI1640 with 0.5% [wt/vol.] BSA and 25 mmol/l HEPES). Chemokines and peptides were prepared in migration assay media and added to the bottom wells of TC-treated six well plates in a total volume of 0.6 ml in duplicate. Input cells (1 × 106) were added to the top of a 5 µm transwell membrane (Corning, Lowell, MA, USA). Where indicated, the transwell membranes were coated overnight with 20 µg/ml fibronectin (BD Biosciences) followed by blocking with 2% (wt/vol.) BSA in PBS. After 4 h, cells that had migrated to the bottom well were collected and counted on a FACSCalibur flow cytometer, using FACS calibrate beads (BD Biosciences) as a normalisation standard. Dead cells were excluded from analysis by the addition of propidium iodide (Sigma-Aldrich).
Apoptosis studies
Primary thymocytes (1 × 106) in complete RPMI media were added to wells of TC-treated round-bottom 96-well plates. For apoptosis rescue experiments, thymocytes were pretreated for 1 h at 37°C with the indicated reagents, following by addition of dexamethasone, anti-Fas antibody or staurosporine. In loss-of-function studies, the aforementioned reagents were added directly to the cells. All treatments were performed in triplicate. Cells were collected 6 h after dexamethasone treatment and 18 h following anti-Fas antibody and staurosporine addition, stained for apoptosis markers using the annexin-V/7-AAD apoptosis detection kit (BD Pharmingen) and analysed on a FACSCalibur flow cytometer.
Statistical analysis
Results are expressed as means±SE, or where indicated means±SD. All statistical analyses were performed with PrismVersion 4.0 software (GraphPAD Software, San Diego, CA, USA). Statistical significance was assessed by one- or two-way ANOVA. Differences were considered statistically significant at p ≤ 0.05.
Results
Glp1r mRNA expression in sorted immune populations
mRNA transcripts for Glp1r were analysed in RNA from sorted immune populations from spleens, thymuses, lymph nodes and bone marrow of C57Bl/6 and normoglycaemic NOD/Ltj mice (Fig. 1). Purity of sorted cells was ≥90% (ESM Figs 1 and 2). Glp1r mRNA transcripts were detected in multiple immune cell populations from NOD mice (Fig. 1b,d). In contrast, Glp1r mRNA transcripts were less abundant in immune cells from the C57 background (Fig. 1a,c). Most notably, Glp1r mRNA levels are present in mature CD4+ and CD8+ T cells isolated from lymph nodes of NOD mice. In comparison, on the C57 background, Glp1r mRNA was detected only in CD4+ T cells from female mice and only in CD8+ T cells from male mice (Fig. 1a–d, lanes 12 and 13). Splenic CD19+ B cells from the NOD but not the C57 background were positive for Glp1r mRNA (Fig. 1a–d, lane 17). Additionally, we examined splenic CD4+CD25+ cells, at least 75% of which also expressed FOXP3 in independent experiments, and are therefore thought to be Tregs [20]. Glp1r mRNA transcripts were expressed in CD4+CD25+ cells isolated from both male and female NOD mice, but were only present in CD4+CD25+ cells from male C57 mice (Fig. 1a–d, lane 18).

Glp1r mRNA expression in immune subsets. Primary thymocytes (lanes 1–5), bone marrow cells (lanes 6–10), lymph node cells (lanes 11–13) and splenocytes (lanes 14–18) from male and female C57Bl/6 (a, c, respectively) and male and female NOD/Ltj mice (b, d, respectively) were stained and sorted on a FACS Aria. RNA was extracted from both the sorted populations and identical cell preparations prior to sorting (total populations) and target cDNA (30 ng RNA equivalent for sorted populations and 112.5 ng RNA equivalent for total populations) was analysed for the expression of mouse Glp1r mRNA. β-Actin mRNA expression was used as a control for the amount of starting RNA. The samples were loaded as follows. Lane 1: unsorted thymocytes; lane 2: CD4+CD8+ thymocytes; lane 3: CD4−CD8– thymocytes; lane 4: CD4+CD8– thymocytes; lane 5: CD4−CD8+ thymocytes; lane 6: unsorted bone marrow cells; lane 7: Gr1 lo/neg, CD11b Hi myeloid progenitors; lane 8: Gr1 Hi, CD11b Hi granulocytes; lane 9: CD19+ cells; lane 10: TCR+ cells; lane 11: unsorted lymph node cells; lane 12: CD4+TCR+ T cells; lane 13: CD8+TCR+ T cells; lane 14: unsorted splenocytes; lane 15: Gr1 lo/neg, CD11b Hi macrophages/monocytes; lane 16: Gr1 Hi, CD11b Hi granulocytes; lane 17: CD19+ cells; lane 18: CD4+CD25+ cells. For each sorted population, cells were pooled from two or three mice, and results were confirmed in two separate experiments
GLP-1R activation leads to cAMP production in primary immune cells
GLP-1R activation is coupled to cAMP accumulation in multiple cell types [21]. When cells isolated from the thymuses, spleens and lymph nodes of NOD mice were treated with 10 or 100 nmol/l of the GLP-1R agonist Ex-4 for 15 min, cAMP production was significantly increased (Fig. 2a–c). Ex-4 also increased cAMP accumulation in primary splenocytes and thymocytes from C57 mice (Fig. 2d,f). In contrast, we could not detect an elevation in cAMP levels in C57 lymph node cells (Fig. 2e). Furthermore, the GLP-1R antagonist Ex(9–39) significantly attenuated cAMP production in response to Ex-4 in primary thymocytes and decreased basal cAMP levels in primary lymph node cells (Fig. 2g,h).

Effects of GLP-1R activation on cAMP production in primary murine immune cells. Single-cell populations from the thymus (a, d, g) and axillary, inguinal, mandibular and superficial parotid and mesenteric lymph nodes (b, e, h) and spleen (c, f, i) of NOD (a–c) and C57 (d–i) mice were treated with the GLP-1R agonist Ex-4 (black triangles), alone (a–f) or in combination with the GLP-1R antagonist Exe(9–39) (white triangles) (g–i). PGE-2 (black circles) was used as a positive control. g–i Exe(9–39) was added 10 min prior to treatment with Ex-4. After 15 min (a–f) or 10 min (g–i) of treatment, cells were lysed and total cAMP levels were quantified by RIA. Results are representative of two independent experiments. All treatments were performed in triplicate. Means±SE. *p < 0.05, **p < 0.01, ***p < 0.001 for treatment vs control (no peptide treatment); † p < 0.05 for GLP-1R agonist Ex-4 in combination with the GLP-1R antagonist Exe(9–39) vs Ex-4
Loss of GLP-1R signalling impairs proliferative responses
Primary thymocytes and lymph node cells from Glp1r +/+ and Glp1r −/− mice were compared for their ability to proliferate in response to mitogens (Fig. 3a,b). Glp1r −/− thymocytes displayed a normal proliferative response to anti-CD3 antibody alone, or in combination with anti-CD28. In contrast, the proliferative response to ConA and to PMA and ionomycin was significantly reduced in Glp1r −/− thymocytes (Fig. 3a). Peripheral lymphocytes from Glp1r −/− mice exhibited an enhanced proliferative response to both ConA and PMA plus ionomycin, as well as to a combination of anti-CD3 and anti-CD28 antibodies (Fig. 3b). Nevertheless, activation of GLP-1R with Ex-4, either alone or in combination with anti-CD3 antibody, did not result in a significantly different proliferation response in both central and peripheral WT lymphocytes (Fig. 3c,d).

GLP-1R activation and proliferation of primary lymphocytes. Primary thymocytes (a) and lymph node cells (b) from female Glp1r +/+ (black bars) and Glp1r −/− (white bars) mice were compared for their ability to proliferate in response to multiple mitogens. Single-cell suspensions from thymuses (c) and lymph nodes (d) of female C57Bl/6 WT were pretreated with a series of mitogens, including the GLP-1R agonist Ex-4, either alone or with an anti-CD3 antibody. Primary thymocytes in (a) and (c) were cultured in the presence of IL-2 (10 U/ml). a, b ConA was used at a concentration of 1 μg/ml, PMA at a concentration of 1 μg/ml and ionomycin at 0.25 μg/ml. Cells were treated with the mitogens for 48 h, followed by addition of [3H]thymidine. Cells were harvested the next day for detection of incorporation of radioactivity, an index of cell proliferation. Data are mean stimulation indices ± SE of triplicate cultures, and are representative of three independent experiments. *p < 0.05, ***p < 0.001 for Glp1r +/+ vs Glp1r −/− cells
GLP-1R signalling is not required for the migration of primary lymphocytes
We next assessed the migratory response of Glp1r −/− thymocytes (Fig. 4a,c) and peripheral lymph nodes cells (Fig. 4b,d) towards increasing concentrations of SDF1a (Fig. 4a,b) and MIP-3b (Fig. 4c,d). No significant differences in migration were observed between the three genotypes. Furthermore, Ex-4 had no significant effects on chemotaxis of immune cells from C57Bl/6 (ESM Fig. 3) and NOD (ESM Fig. 4) mice, either alone or in combination with the chemokines SDF1a, MIP-3b or CCL25. In addition, pre-treatment with Ex-4 for 30 min had no effect on the migratory behaviour of primary thymocytes and splenocytes (ESM Fig. 4c,h).

GLP-1R signalling is not required for the migration of primary lymphocytes. Primary thymocytes (a, c) and cells from peripheral lymph nodes (b, d) from Glp1r +/+ (black bars), Glp1r +/− (grey bars) and Glp1r −/− (white bars) mice were compared for their ability to migrate in response to increasing concentrations of SDF1a (a, b), or MIP-3b (c, d). Cells were placed on the top well of a fibronectin-coated transwell insert. On the bottom well, media (control) or media with chemokines was added. After a 4 h incubation, cells that had migrated to the bottom well were collected and counted on a FACSCalibur. Results are means±SE (n = 3–6 mice per genotype). All treatments were performed in duplicate
GLP-1R signalling does not modify cell death in murine thymocytes
There were no significant differences in thymocyte apoptosis following exposure of Glp1r −/− vs Glp1r +/+ cells to dexamethasone (Fig. 5a), staurosporine (Fig. 5b) or anti-Fas antibody (Fig. 5c). Similarly, Ex-4 failed to modify apoptosis in primary thymocytes isolated from C57 and NOD mice (ESM Fig. 5), while pre-treatment with either cycloheximide or IL-4 reduced thymocyte apoptosis following exposure to dexamethasone (ESM Fig. 5a,b).

GLP-1R signalling does not modify cell survival in primary thymocytes. Primary thymocytes from Glp1r +/+ (black bars), Glp1r +/− (grey bars) and Glp1r −/− (white bars) mice were compared for their ability to survive apoptosis in response to 100 nmol/l dexamethasone (a), 1 µmol/l staurosporine (b) or activation of the Fas receptor with 1 µg/ml anti-Fas antibody (aFas) (c). Apoptosis was measured using annexin-V and the viability dye 7-AAD on a FACSCalibur. The percentage of apoptotic cells (annexin-V positive/7-AAD negative) is shown. Data are means±SE (n = 3–4 mice per genotype)
Lymphocyte subsets in Glp1r−/− mice
Lymphocyte subsets from the bone marrow, thymus, spleen and lymph nodes of young (<12 weeks) and older (>12 weeks of age) male and female Glp1r −/− mice and Glp1r +/+ littermate control mice were quantified (ESM Figs 6, 7, 8, 9, 10, 11, 12 and 13). The number of cells in lymph nodes was significantly greater in older female Glp1r −/− mice (Fig. 6c). There were no genotype differences in organ cellularity in younger males and females (Fig. 6a,b) and older males (Fig. 6d).

Lymphoid cellularity in Glp1r −/− mice. Spleens, lymph nodes (axillary, inguinal, mandibular and superficial parotid and mesenteric), thymuses and the bone marrow (BM) from both tibia and femur were isolated from mice with a null mutation in the Glp1r gene (Glp1r −/−) (white bars) and littermate controls (Glp1r +/+ [black bars], Glp1r +/− [grey bars]). Organ cellularity was determined for female (a) and male (b) mice <12 weeks of age, and female (c) and male (d) mice >12 weeks of age. Data are means ± SE (n = 2–5 mice per genotype). *p < 0.05 for Glp1r −/− vs Glp1r +/+ cells
A significant reduction in the percentage of peripheral CD4+CD25+FOXP3+ Tregs was observed in cells from lymph nodes of young male Glp1r −/− mice (Fig. 7h). However, this difference was not observed in Tregs isolated from the spleen (Fig. 7k) or in peripheral Tregs from young female mice (Fig. 8i,l). In the thymus of young female mice, the percentage of immature CD4− CD8− double-negative cells was significantly higher and the percentage of CD4+ CD8+ double-positive cells was significantly lower in mice with one functional copy of the Glp1r vs WT and knockout animals (Fig. 8d,e). However, these differences in immature T cells were not seen in the periphery, as the proportion of peripheral T cells in young female Glp1r −/− mice were similar to those of littermate controls (Fig. 8h,k). In addition, the percentage of peripheral CD4+ CD8+ double-positive T cells in both the lymph nodes and the spleen was not different in the three genotypes (data not shown). Older female Glp1r −/− mice had a significantly higher percentage of immature B220+IgM− B cells in their bone marrow compared with Glp1r +/+ mice (ESM Fig. 12b). Nevertheless, this difference was not persistent when we looked at the percentages of both immature and mature B cells in the periphery (ESM Fig. 12g,i). Similarly, the percentages of both mature and immature B and T cells in male mice >12 weeks was comparable between the three genotypes (ESM Fig. 13). When data were expressed as absolute numbers, no significant differences were observed between the three genotypes in both young and older mice (ESM Tables 2, 3, 4 and 5).

Immunophenotyping analysis of male Glp1r −/− mice. Single-cell suspensions from bone marrow (a, b), thymus (c–e), lymph nodes (f–h) and spleen (i–k) of Glp1r −/− (white bars) and littermate control (Glp1r +/+ [black bars], Glp1r +/− [grey bars]) male mice <12 weeks of age, were analysed for the percentage of different immune subsets as shown, using flow cytometry. Dead cells (propidium iodide-positive cells) were excluded from analysis. Data are means±SE (n = 2–4 mice per genotype). *p < 0.05 for Glp1r −/− vs Glp1r +/− and Glp1r +/+ cells

Immunophenotyping analysis of female Glp1r −/− mice. Single-cell suspensions from bone marrow (a–c), thymuses (d–f), lymph nodes (g–i) and spleen (j–l) of Glp1r +/+ (black bars), Glp1r +/− (grey bars) and Glp1r −/− (white bars) female mice (<12 weeks of age) were analysed for the percentage of different immune subsets as shown, using flow cytometry. Results are plotted as a percentage of total live cells and shown as means ± SE (n = 2–5 mice per genotype). *p < 0.05 for Glp1r −/− vs Glp1r +/− cells; ***p < 0.001 for Glp1r +/− vs Glp1r +/+ and Glp1r −/−
Discussion
GLP-1R agonists and dipeptidyl peptidase-4 inhibitors are approved for the treatment of type 2 diabetes [22] and are being examined in clinical trials of islet transplantation in patients with type 1 diabetes [16, 17], an autoimmune disorder resulting from defective immunoregulation [23, 24]. A subset of patients with type 2 diabetes may also exhibit a dysregulated immune system [25–27]. Previous work from our laboratory [12], as well as others [15] suggested that GLP-1R signalling may regulate T cell subsets, including Tregs. We have now assessed the effects of activating or eliminating the GLP-1R in subsets of murine immune cells.
Although GLP-1 action was first localised to beta cells [28], the GLP-1R is detected in extrapancreatic tissues, including liver, lung, kidney, heart, brain and intestine [29, 30]. We previously demonstrated that Glp1r transcripts are expressed in spleen, thymus and lymph nodes of NOD and C57Bl/6 mice [12]. Our current findings illustrate that the Glp1r is more widely expressed in immune subpopulations from the NOD, relative to the C57BL/6 background. Chiu et al. demonstrated a higher frequency of activated B and T cells in the NOD mouse compared with other strains, including the C57BL/6 mouse [31]. Hence one potential explanation for the differential Glp1r expression could be the activation status of lymphocytes, as we did not discriminate between rested and activated B and T cells during the sorting procedure. Further studies are required to examine the significance of and mechanisms underlying the differential Glp1r expression on the NOD vs C57 background.
Intriguingly, there were sex-specific differences in Glp1r expression. There are no reports examining sex differences in GLP-1R abundance in immune cells, or in other tissues. Perhaps not surprisingly there are reported sex-specific patterns of gene expression in the NOD mouse [32]. Hence the observed differences in GLP-1R abundance may reflect both species-specific and sex-specific differences in the control of GLP-1R production. As diabetes development demonstrates a strong sex bias in the NOD mouse, the implications arising from differences in GLP-1R levels require further examination.
Our analyses reveal that several immature lymphocyte subsets in the thymus and bone marrow express Glp1r transcripts. In particular, CD19+ B cells from the bone marrow of female but not male C57 mice are positive for Glp1r transcripts. Moreover, aged female Glp1r −/− mice have a significantly increased percentage of immature B220+IgM− B cells in their bone marrow, implicating GLP-1R signalling in B cell maturation. Further studies are required to determine the timing and significance of GLP-1R production during haematopoiesis and lymphocyte maturation.
Glp1r transcripts were also detected in sorted CD4+CD25+ cells, a cell subset mostly comprising Tregs [33]. Glp1r transcripts are present in CD4+CD25+ cells isolated from the spleens of male C57 mice. Immunophenotyping analysis revealed a significant deficiency in the percentage of CD4+CD25+FOXP3+ Tregs from the lymph nodes of male Glp1r −/− mice, thus suggesting that GLP-1R signalling could play a role in the maintenance and functioning of Tregs. Conversely, we were not able to detect Glp1r mRNA transcripts in splenic samples of female C57 mice, and we did not find a difference in the percentage of splenic and lymph node Tregs of female Glp1r −/− mice.
On the NOD background, CD4+CD25+ cells from both male and female mice were positive for Glp1r transcripts. Very little is known about a potential role for GLP-1R in the frequency and suppressive capacity of Tregs in the setting of an autoimmune attack. Previous work from our laboratory demonstrated a decrease in the number of thymic Tregs in NOD mice receiving chronic Ex-4 treatment, although we did not detect any differences in peripheral Tregs isolated from lymph nodes [12]. Intriguingly, Suarez-Pinzon et al. have shown that treatment of diabetic NOD mice with a combination of GLP-1 and gastrin results in a diminished autoimmune response during the transplantation of syngeneic islets. The authors observed an induction of suppressive cytokine secretion in islet-infiltrating leucocytes, and concluded that the GLP-1 and gastrin combination therapy may regulate immune pathways in the NOD mice [13]. A recent study investigated the role of GLP-1R activation in the number and function of Tregs in NOD mice. Treatment with 200 ng Ex-4 for 30 days increased the number of splenic Tregs. Furthermore, a trend towards enhanced Treg function was observed in CD4+CD25+ splenocytes isolated from Ex-4-treated mice [15].
Our data demonstrated that GLP-1R activation leads to a modest but significant increase in cAMP accumulation in mixed leucocyte populations. Elevated cAMP has been shown to be a mechanism by which Tregs induce immunosuppression. In fact, Tregs express high levels of cAMP, and are able to induce cAMP accumulation in activated target cells by multiple mechanisms [34, 35]. Taking into consideration that GLP-1R activation modulates Treg function [15], it is possible to speculate that this mechanism is cAMP dependent. Further experiments are required to elucidate potential mechanism(s) by which GLP-1R activation augments Treg function.
GLP-1R activation did not enhance proliferation of primary thymocytes and lymph node cells, either alone or as a costimulatory signal when combined with anti-CD3. In contrast, loss of GLP-1R signalling in primary thymocytes resulted in a significantly diminished proliferation response upon ConA and PMA plus ionomycin stimulation, but there was no difference on the proliferative response when thymocytes were stimulated with anti-CD-3 and anti-CD-3 plus anti-CD-28 activation. Stimulation of T cells with ConA is dependent on changes in cellular cAMP [36, 37]. Hence it is possible that GLP-1R activation has an additive effect on cAMP accumulation following activation with ConA, and the absence of this additive effect, as is the case in immune cells lacking functional GLP-1R, results in a defective proliferative response.
The combination of PMA and ionomycin induces proliferation by directly activating protein kinase C (PKC) and increasing intracellular calcium levels, hence bypassing T cell reception activation [38]. The fact that thymocytes from Glp1r −/− mice displayed a hypoproliferative response to PMA plus ionomycin suggests that GLP-1R signalling is required subsequent to activation of PKC. Potential candidates could be transcription factors, as GLP-1R activation in the islet beta cells has been shown to promote nuclear translocation of nuclear factor kappaB and the nuclear factor of activated T cells, and to activate several immediate early-response genes and proto-oncogenes [39–41], which are common in the signalling pathway of TCR activation [42].
Paradoxically, Glp1r −/− peripheral lymph node cells exhibited a significantly enhanced proliferative response to mitogens. It has been shown that in mouse islets, genetic disruption of GLP-1R signalling results in compensatory changes in signalling pathways. In the study by Flamez et al., islets lacking a functional GLP-1R also demonstrated elevated cytosolic calcium levels, and this increase in calcium was observed at a significantly lower threshold for glucose concentration [43]. These results might partially explain the hyperresponsiveness of immune cells lacking GLP-1R signalling, as elevation in cytosolic calcium is a triggering signal for lymphocyte proliferation (as reviewed [44]).
The hypoproliferative responses in thymocytes from Glp1r −/− mice were observed only when thymocytes were stimulated with non-physiological mitogens (PMA and ConA). Hence it seems that GLP-1R signalling is not critical for the endogenous proliferation of thymocytes. However, we did observe a defect in response to anti-CD3 plus anti-CD28 in peripheral lymphocytes, which suggests that GLP-1R signalling may be required for optimal proliferation of peripheral lymphocytes under these experimental conditions. A potential explanation for these differences could be that, in the thymus, absent GLP-1R signalling is compensated by upregulation of other mechanisms not operative in the periphery. Indeed, it was previously reported that glucoregulatory actions of GLP-1 are compensated via other mechanisms in Glp1r −/− mice [45].
GLP-1R signalling in immune cells was not associated with modulation of anti-apoptotic pathways. In contrast, GLP-1R activation inhibits apoptosis in other cell types including islet beta cells, cardiomyocytes and neurons [46–48]. However, we cannot exclude the possibility that a modest effect of GLP-1 on cell survival may have been obscured, as the importance of GLP-1R signalling for cytoprotection was studied in mixed immune populations. Hence, further viability studies are required in isolated immune subsets producing the GLP-1R to resolve whether GLP-1R signalling has a role in leucocyte apoptosis.
Similarly, we were not able to detect any effects on lymphocyte migration upon either GLP-1R activation or loss of GP-1R signalling in mixed primary immune populations. This result was also unanticipated, as it is well accepted that G protein-coupled receptor signalling is crucial for chemotaxis [49, 50]. The GLP-1R, although primarily coupled to Gαs, has also been shown to couple to the Gαi protein [51], which is essential for the migration response. In agreement with our results, Kim et al. also did not find a significant effect on migration of GLP-1-treated splenic CD4+ T cells [52].
In conclusion, we report that Glp1r mRNA is widely expressed in several immune subpopulations. Activation of the GLP-1R leads to cAMP accumulation, and GLP-1R signalling contributes to the regulation of both thymocyte and peripheral T cell proliferation. Moreover, the GLP-1R is involved in the maintenance of peripheral Tregs. These findings expand our understanding of the role of the GLP-1R in the development, maintenance and function of the immune system.
Abbreviations
- 7-AAD:
-
7-Amino-actinomycin D
- B220:
-
CD45R/protein tyrosine phosphatase, receptor type, C
- ConA:
-
Concanavalin A
- Ex-4:
-
Exendin-4
- FOXP3:
-
Forkhead box protein 3
- GLP-1:
-
Glucagon-like peptide-1
- GLP-1R:
-
Glucagon-like peptide-1 receptor
- MIP-3b:
-
Macrophage inflammatory protein 3b
- PGE-2:
-
Prostaglandin E2
- PMA:
-
Phorbol myristate acetate
- SDF1a:
-
Stromal cell-derived factor 1a
- TC:
-
Tissue culture
- TCR:
-
T cell receptor
- Treg:
-
Regulatory T cell
- WT:
-
Wild-type
References
Madsbad S, Krarup T, Deacon CF, Holst JJ (2008) Glucagon-like peptide receptor agonists and dipeptidyl peptidase-4 inhibitors in the treatment of diabetes: a review of clinical trials. Curr Opin Clin Nutr Metab Care 11:491–499
Dupre J (2005) Glycaemic effects of incretins in type 1 diabetes mellitus: a concise review, with emphasis on studies in humans. Regul Pept 128:149–157
Sherry NA, Chen W, Kushner JA et al (2007) Exendin-4 improves reversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody by enhancing recovery of beta-cells. Endocrinology 148:5136–5144
Ogawa N, List JF, Habener JF, Maki T (2004) Cure of overt diabetes in NOD mice by transient treatment with anti-lymphocyte serum and exendin-4. Diabetes 53:1700–1705
Yang Z, Chen M, Carter JD et al (2006) Combined treatment with lisofylline and exendin-4 reverses autoimmune diabetes. Biochem Biophys Res Commun 344:1017–1022
Tian B, Hao J, Zhang Y et al (2009) Upregulating CD4+CD25+FOXP3+ regulatory T cells in pancreatic lymph nodes in diabetic NOD mice by adjuvant immunotherapy. Transplantation 87:198–206
National Institutes of Health Clinical Center (2009) Effect of AC2993 with or without immunosuppression on beta cell function in patients with type I diabetes. Available from www.clinicaltrials.gov/, accessed 6 September 2009.
Toyoda K, Okitsu T, Yamane S et al (2008) GLP-1 receptor signaling protects pancreatic beta cells in intraportal islet transplant by inhibiting apoptosis. Biochem Biophys Res Commun 367:793–798
King A, Lock J, Xu G, Bonner-Weir S, Weir GC (2005) Islet transplantation outcomes in mice are better with fresh islets and exendin-4 treatment. Diabetologia 48:2074–2079
Juang JH, Kuo CH, Wu CH, Juang C (2008) Exendin-4 treatment expands graft beta-cell mass in diabetic mice transplanted with a marginal number of fresh islets. Cell Transplant 17:641–647
Zhang J, Tokui Y, Yamagata K et al (2007) Continuous stimulation of human glucagon-like peptide-1 (7–36) amide in a mouse model (NOD) delays onset of autoimmune type 1 diabetes. Diabetologia 50:1900–1909
Hadjiyanni I, Baggio LL, Poussier P, Drucker DJ (2008) Exendin-4 modulates diabetes onset in nonobese diabetic mice. Endocrinology 149:1338–1349
Suarez-Pinzon WL, Power RF, Yan Y, Wasserfall C, Atkinson M, Rabinovitch A (2008) Combination therapy with glucagon-like peptide-1 and gastrin restores normoglycemia in diabetic NOD mice. Diabetes 57:3281–3288
Xue S, Wasserfall C, Parker M et al (2009) Exendin-4 treatment of nonobese diabetic mice increases beta-cell proliferation and fractional insulin reactive area. J Diabetes Complicat. doi:10.1016/j.jdiacomp.2008.12.004
Xue S, Wasserfall CH, Parker M et al (2008) Exendin-4 therapy in NOD mice with new-onset diabetes increases regulatory T cell frequency. Ann N Y Acad Sci 1150:152–156
Ghofaili KA, Fung M, Ao Z et al (2007) Effect of exenatide on beta cell function after islet transplantation in type 1 diabetes. Transplantation 83:24–28
Froud T, Faradji RN, Pileggi A et al (2008) The use of exenatide in islet transplant recipients with chronic allograft dysfunction: safety, efficacy, and metabolic effects. Transplantation 86:36–45
Poussier P, Ning T, Murphy T, Dabrowski D, Ramanathan S (2005) Impaired post-thymic development of regulatory CD4+25+ T cells contributes to diabetes pathogenesis in BB rats. J Immunol 174:4081–4089
Vielkind S, Gallagher-Gambarelli M, Gomez M, Hinton HJ, Cantrell DA (2005) Integrin regulation by RhoA in thymocytes. J Immunol 175:350–357
Fontenot JD, Gavin MA, Rudensky AY (2003) Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4:330–336
Doyle ME, Egan JM (2007) Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther 113:546–593
Lovshin JA, Drucker DJ (2009) Incretin-based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol 5:262–269
Morran MP, Omenn GS, Pietropaolo M (2008) Immunology and genetics of type 1 diabetes. Mt Sinai J Med 75:314–327
Tisch R, Wang B (2008) Dysregulation of T cell peripheral tolerance in type 1 diabetes. Adv Immunol 100:125–149
Donath MY, Ehses JA, Maedler K et al (2005) Mechanisms of beta-cell death in type 2 diabetes. Diabetes 54(Suppl 2):S108–S113
Ehses JA, Boni-Schnetzler M, Faulenbach M, Donath MY (2008) Macrophages, cytokines and beta-cell death in Type 2 diabetes. Biochem Soc Trans 36:340–342
Guest CB, Park MJ, Johnson DR, Freund GG (2008) The implication of proinflammatory cytokines in type 2 diabetes. Front Biosci 13:5187–5194
Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF (1987) Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci U S A 84:3434–3438
Campos RV, Lee YC, Drucker DJ (1994) Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology 134:2156–2164
Bullock BP, Heller RS, Habener JF (1996) Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology 137:2968–2978
Chiu PP, Jevnikar AM, Danska JS (2001) Genetic control of T and B lymphocyte activation in nonobese diabetic mice. J Immunol 167:7169–7179
Ivakine EA, Fox CJ, Paterson AD et al (2005) Sex-specific effect of insulin-dependent diabetes 4 on regulation of diabetes pathogenesis in the nonobese diabetic mouse. J Immunol 174:7129–7140
Sgouroudis E, Piccirillo CA (2009) Control of type 1 diabetes by CD4+Foxp3+ regulatory T cells: lessons from mouse models and implications for human disease. Diabetes Metab Res Rev 25:208–218
Sojka DK, Huang YH, Fowell DJ (2008) Mechanisms of regulatory T cell suppression: a diverse arsenal for a moving target. Immunology 124:13–22
Vendetti S, Patrizio M, Riccomi A, de Magistris MT (2006) Human CD4+ T lymphocytes with increased intracellular cAMP levels exert regulatory functions by releasing extracellular cAMP. J Leukoc Biol 80:880–888
Wang T, Sheppard JR, Foker JE (1978) Rise and fall of cyclic AMP required for onset of lymphocyte DNA synthesis. Science 201:155–157
Takigawa M, Waksman BH (1981) Mechanisms of lymphocyte ‘deletion’ by high concentrations of ligand. I. Cyclic AMP levels and cell death in T-lymphocytes exposed to high concentrations of concanavalin A. Cell Immunol 58:29–38
Truneh A, Albert F, Golstein P, Schmitt-Verhulst AM (1985) Early steps of lymphocyte activation bypassed by synergy between calcium ionophores and phorbol ester. Nature 313:318–320
Buteau J, El-Assaad W, Rhodes CJ, Rosenberg L, Joly E, Prentki M (2004) Glucagon-like peptide-1 prevents beta cell glucolipotoxicity. Diabetologia 47:806–815
Susini S, Roche E, Prentki M, Schlegel W (1998) Glucose and glucoincretin peptides synergize to induce c-fos, c-jun, junB, zif-268, and nur-77 gene expression in pancreatic beta(INS-1) cells. FASEB J 12:1173–1182
Lawrence MC, Bhatt HS, Easom RA (2002) NFAT regulates insulin gene promoter activity in response to synergistic pathways induced by glucose and glucagon-like peptide-1. Diabetes 51:691–698
Song J, Lei FT, Xiong X, Haque R (2008) Intracellular signals of T cell costimulation. Cell Mol Immunol 5:239–247
Flamez D, Gilon P, Moens K et al (1999) Altered cAMP and Ca2+ signaling in mouse pancreatic islets with glucagon-like peptide-1 receptor null phenotype. Diabetes 48:1979–1986
Lewis RS (2001) Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol 19:497–521
Pederson RA, Satkunarajah M, McIntosh CH et al (1998) Enhanced glucose-dependent insulinotropic polypeptide secretion and insulinotropic action in glucagon-like peptide 1 receptor −/− mice. Diabetes 47:1046–1052
Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ (2003) Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. J Biol Chem 278:471–478
Noyan-Ashraf MH, Momen MA, Ban K et al (2009) GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 58:975–983
Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH (2002) Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther 302:881–888
Cinalli RM, Herman CE, Lew BO, Wieman HL, Thompson CB, Rathmell JC (2005) T cell homeostasis requires G protein-coupled receptor-mediated access to trophic signals that promote growth and inhibit chemotaxis. Eur J Immunol 35:786–795
Moser B, Loetscher P (2001) Lymphocyte traffic control by chemokines. Nat Immunol 2:123–128
Hallbrink M, Holmqvist T, Olsson M, Ostenson CG, Efendic S, Langel U (2001) Different domains in the third intracellular loop of the GLP-1 receptor are responsible for Galpha(s) and Galpha(i)/Galpha(o) activation. Biochim Biophys Acta 1546:79–86
Kim SJ, Nian C, Doudet DJ, McIntosh CH (2009) Dipeptidyl peptidase IV inhibition with MK0431 improves islet graft survival in diabetic NOD mice partially via T cell modulation. Diabetes 58:641–651
Acknowledgements
The authors would like to thank A. Wong for assistance with the immunophenotyping experiments, B. Dong for assistance with the proliferation, apoptosis and migration experiments, D. Holland for excellent technical assistance and J. Zhang for helpful discussions on data analysis and interpretation. In addition, the authors would like to thank the staff at the SickKids-UHN flow cytometry facility (Toronto, ON, Canada), in particular S. Zhao, P.-A. Penttilä, L. Jamieson, M. Tseng and A. Bang from Mt Sinai Flow Cytometry facility for their expertise in cell sorting. These studies were supported in part by the Canada Research Chairs Program, and a grant from the Juvenile Diabetes Association (1-2006-696) and the Canadian Research Chairs Program to D. J. Drucker.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM Table 1
Primers used for detection of Glp1r expression in sorted immune cells (PDF 43 kb)
ESM Table 2
Absolute cell numbers in immune subpopulations from the bone marrow, thymus, lymph nodes and spleen of young male mice (PDF 26 kb)
ESM Table 3
Absolute cell numbers in immune subpopulations from the bone marrow, thymus, lymph nodes and spleen of young female mice (PDF 45 kb)
ESM Table 4
Absolute cell numbers in immune subpopulations from the bone marrow, thymus, lymph nodes and spleen of female mice aged 12 weeks or older (PDF 25 kb)
ESM Table 5
Absolute cell numbers in immune subpopulations from bone marrow, thymus, lymph nodes and spleen of male mice aged 12 weeks and older (PDF 20.4 KB)
ESM Fig. 1
Pre- and post-sort analysis of primary thymocytes and bone marrow cells. Primary cells isolated from the thymus (A) or bone marrow (B) of male NOD mice were stained and sorted on a FACS Aria. Two-variable 5% probability contour plots indicate the percentages of each population prior to sorting, while dot plots indicate purity subsequent to sorting (PDF 65 kb)
ESM Fig. 2
Pre- and post-sort analysis of peripheral immune subpopulations. Representative plots of pre- and post-sorted primary lymph node cells (A) or splenocytes (B) isolated from male NOD mice. The frequencies of the indicated subpopulations are shown (PDF 61 kb)
ESM Fig. 3
GLP-1R signalling is not required for the migration of primary lymphocytes: gain-of-function studies. Single-cell suspensions from thymuses (A–C), peripheral lymph nodes (D–F), mesenteric lymph nodes (G–I) and spleens (J–L) of female C57Bl/6 were allowed to migrate towards increasing doses of the GLP-1R agonist Ex-4, either alone or in combination with the indicated chemokines. Results are displayed as means ± SE of three to six independent experiments. Every treatment was performed in duplicate (PDF 62 kb)
ESM Fig. 4
GLP-1R signalling is not required for the migration of primary NOD lymphocytes. Single-cell suspensions from thymuses (A–C), peripheral lymph nodes (D, E) and spleens (F–H) of female normoglycaemic NOD/Ltj mice were allowed to migrate towards increasing doses of the GLP-1R agonist Ex-4, either alone (A, D, F) or in combination with SDF1a (B, E, G). In a different set of experiments, cells were pretreated with Ex-4 for 30 min, and were then allowed to migrate towards the chemokine SDF1a (C, H). Results are displayed as means ± SE of three to six independent experiments. Every treatment was performed in duplicate (PDF 48 kb)
ESM Fig. 5
GLP-1R signalling does not increase survival of primary thymocytes: gain-of-function studies. Single-cell suspensions from thymuses of female C57Bl/6 (A, C, E) and female normoglycaemic NOD/Ltj mice (B, D, F) were pretreated with increasing doses of the GLP-1R agonist Ex-4 (1 nmol/l Ex-4, light-grey bars; 10 nmol/l Ex-4, medium-grey bars; 100 nmol/l Ex-4 dark-grey bars) for 1 h, followed by addition of 100 nmol/l dexamethasone (A, B), 1 µmol/l staurosporine (C, D) or 1 µg/ml anti-Fas (aFas) antibody (E, F) to induce apoptosis. IL-4 (hatched bars) was used at a concentration of 10 ng/ml, forskolin (cross-hatched bars) at 10 µmol/l, PGE-2 (horizontal-striped bars) at 100 nmol/l and cycloheximide (dotted bars) at 10 μg/ml. Cells without any treatment are shown as white bars. Cells were then stained with annexin-V and 7-AAD, and apoptosis was quantified using the FACSCalibur flow cytometer. The percentages of apoptotic cells (annexin-V positive/7-AAD negative) are shown. Data are means ± SE. All treatments were performed in duplicate. n = 2–3 independent experiments. *p<0.05, **p<0.01 and ***p<0.001 treatment vs control (PDF 59 kb)
ESM Fig. 6
Phenotypic profile of bone marrow cells and thymocytes of male Glp1r −/− mice. Single-cell suspensions from bone marrow (A, B) and thymuses of Glp1r +/+, Glp1r +/− and Glp1r −/− male mice (younger than 12 weeks) were analysed for the percentage of different immune subsets as indicated. Dead cells (propidium iodide-positive cells) were excluded from analysis. (A) Results are depicted in a histogram. (B, C) Data are presented in two-variable 5% probability contour plots. Data are means ± SD. n = 2–4 mice/genotype (PDF 51 kb)
ESM Fig. 7
Phenotypic profile of lymph node cells and splenocytes of male Glp1r −/− mice. Phenotypic profile of lymph node cells (A, B) and splenocytes (C, D) isolated from male mice (younger than 12 weeks). 5% probability contour plots (A, B, D) and histogram plots (C) indicate the frequencies of each subpopulation. Data are means ± SD. n = 2–4 mice/genotype (PDF 58 kb)
ESM Fig. 8
Quantification of Tregs in lymph nodes and spleen of male Glp1r −/− mice. Two-variable 2% probability contour plots depict surface expression of CD4 vs CD25 on lymph node cells (A) and splenocytes (C) from male Glp1r −/− mice and littermate controls, aged 12 weeks or younger. Histograms show the intracellular expression of FOXP3 in gated CD4+CD25+ cells from the lymph nodes (B) and spleen (D). Numbers indicate mean frequencies ± SD. n = 2–4 mice/genotype (PDF 28.7 KB)
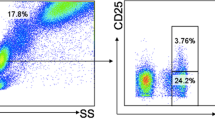
ESM Fig. 9
Phenotypic profile of bone marrow cells and thymocytes of female Glp1r −/− mice. Single-cell suspensions from bone marrow (A, B) and thymuses of Glp1r +/+, Glp1r +/− and Glp1r −/− female mice (younger than 12 weeks) were analysed for the percentage of different immune subsets as indicated. Dead cells (propidium iodide-positive cells) were excluded from analysis. (A) Results are depicted as a two-variable 2% probability contour plots. (B, C) Data are 5% probability contour plots. Data are means ± SD. n = 2–5 mice/genotype (PDF 53 kb)
ESM Fig. 10
Phenotypic profile of lymph node cells and splenocytes of female Glp1r −/− mice. Phenotypic profile of lymph node cells (A, B) and splenocytes (C, D) isolated from female mice (younger than 12 weeks). 5% probability contour plots (A, B, D) and histogram plots (C) indicate the frequencies of each subpopulation. Data are means ± SD. n = 2–5 mice/genotype (PDF 57 kb)
ESM Fig. 11
Quantification of Tregs in lymph nodes and spleen of male Glp1r −/− mice. Two-variable 2% probability contour plots depict surface expression of CD4 vs CD25 on lymph node cells (A) and splenocytes (C) from female Glp1r −/− mice and littermate controls, aged 12 weeks or younger. Histograms show the intracellular expression of FOXP3 in gated CD4+CD25+ cells from the lymph nodes (B) and spleen (D). Numbers indicate mean frequencies ± SD. n = 2–5 mice/genotype (PDF 24.7 KB)
ESM Fig. 12
Immunophenotyping analysis of older female Glp1r −/− mice. Single-cell suspensions from bone marrow (A–C), thymuses (D–F), lymph nodes (G, H) and spleens (I, J) of Glp1r +/+ (black bars), Glp1r +/− (grey bars) and Glp1r −/− (white bars) female mice (12 weeks and older) were analysed for the percentage of different immune subsets as shown, using flow cytometry. Dead cells (propidium iodide-positive cells) were excluded from analysis. Data are means ± SE. n = 2–4 mice/genotype. *p<0.05 Glp1r −/− vs Glp1r +/+ cells (PDF 45 kb)
ESM Fig. 13
Immunophenotyping analysis of older male Glp1r −/− mice. Single-cell suspensions from bone marrow (A–C), thymuses (D–F), lymph nodes (G, H) and spleens (I, J) of Glp1r +/+ (black bars), Glp1r +/− (grey bars) and Glp1r −/− (white bars) male mice (12 weeks and older) were analysed for the percentage of different immune subsets as shown, using flow cytometry. Dead cells (propidium iodide-positive cells) were excluded from analysis. Data are means ± SE. n = 3–5 mice/genotype (PDF 45 kb)
Rights and permissions
About this article
Cite this article
Hadjiyanni, I., Siminovitch, K.A., Danska, J.S. et al. Glucagon-like peptide-1 receptor signalling selectively regulates murine lymphocyte proliferation and maintenance of peripheral regulatory T cells. Diabetologia 53, 730–740 (2010). https://doi.org/10.1007/s00125-009-1643-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1643-x