
Abstract
NKG2D is one of the most intensively studied immune receptors of the past decade. Its unique binding and signaling properties, expression pattern, and functions have been attracting much interest within the field due to its potent antiviral and anti-tumor properties. As an activating receptor, NKG2D is expressed on cells of the innate and adaptive immune system. It recognizes stress-induced MHC class I-like ligands and acts as a molecular sensor for cells jeopardized by viral infections or DNA damage. Although the activating functions of NKG2D have been well documented, recent analysis of NKG2D-deficient mice suggests that this receptor may have a regulatory role during NK cell development. In this review, we will revisit known aspects of NKG2D functions and present new insights in the proposed influence of this molecule on hematopoietic differentiation.
Similar content being viewed by others

The NKG2D receptor and its ligands
NKG2D is a potent activating receptor expressed on virtually all NK cells [1]. It is also expressed on most NKT cells and subpopulations of γδ T cells [2]. All human CD8+ αβ T cells express NKG2D, whereas NKG2D expression on CD4+ αβ T cells has primarily been reported in some pathological conditions [3–6]. Mice express NKG2D only on activated and memory type αβ CD8+ T cells. Thus, NKG2D is expressed on effector cells of both innate and adaptive immune responses and is implicated in the surveillance of viral infections and cancers as well as in some autoimmune processes and transplantation reactions [7–15].
NKG2D is a C-type lectin-like type receptor and belongs to the NK group 2 (NKG2) of receptors as member D. It has also been classified as killer cell lectin-like receptor of the subfamily K, member 1 (KLRK1) [16]. NKG2D (KLRK1) is encoded by the Nkg2d (Klrk1) gene that is located within the NK gene complex (NKC) situated on chromosome 6 in mice and on chromosome 12 in humans [17, 18]. NKG2D is atypical in comparison with other NK receptors. Although NKG2D belongs to the NKG2 family, it does not share most of their properties. In contrast to other members of the family (NKG2A/B, NKG2C, and NKG2E), which form heterodimers with CD94 and recognize MHC class Ib molecules (HLA-E in humans and Qa1 in mouse), NKG2D is a homodimer and recognizes a number of stress induced MHC class I-like ligands [2, 19]. There is no inhibitory counterpart known for NKG2D and NKG2D is capable of overriding signals provided by inhibitory receptors on NK cells engaging MHC class Ia and Ib molecules. Thus, NKG2D plays a role as a molecular sensor detecting “induced self” on cells in danger, which is mostly triggered by viral infections and by factors causing DNA damage and tumor transformation [19, 20].
The NKG2D receptor consists of two disulfide-linked type II transmembrane proteins with positively charged amino acid residues in their transmembrane domains and very short intracellular tails that do not have any signaling properties [21]. Signal transduction operates through two adaptor proteins, DAP10 and DAP12, which associate with the receptor as homodimers [22, 23]. The NKG2D signaling complex appears as a hexameric structure since each NKG2D homodimer binds two adaptor homodimers [24]. In mice, there are two NKG2D isoforms that differ in length of their intracellular sequence by 13 amino acid residues and have different association properties for the two adaptor proteins. The short isoform of NKG2D (NKG2D-S) associates with both adaptor proteins, while the long one (NKG2D-L) only binds to DAP10 [22, 23, 25]. Humans possess only the NKG2D-L isoform.
DAP10 has a YINM motif in its cytoplasmatic tail which, upon phosphorylation, recruits the p85 subunit of phoshpoinositide-3-kinase (PI3K) and Grb2–Vav1. This signal stimulates survival and cytotoxicity of NK cells and provides co-stimulation to activated T cells [26–28]. DAP12 possesses an immunoreceptor tyrosine-based activation motif (ITAM) which, after phosphorylation, recruits Src family kinases ZAP70 and Syk, responsible for cytokine release and enhancement of cytotoxicity in NK cells [29]. Even though there are some reports that DAP12 can also be expressed by T cells [30], deficiency of DAP10 results in complete loss of NKG2D signaling in T cells and therefore appears to be the most important adaptor for NKG2D signaling in these cells [13, 22, 23].
NKG2D is able to bind a number of MHC class I-like ligands because of its unique protein structure [31–33]. In mice, it binds to the family of retinoic acid early inducible proteins (Rae1) [34, 35]. There are five members of these GPI-anchored proteins (Rae1-α, -β, -γ, -δ, and -ε) and their expression differs between mouse strains [19]. NKG2D also binds to the closely related group of histocompatibility antigen 60 (H60) glycoproteins, which consists of three members (H60a–c). H60a and b are transmembrane proteins, while H60c is GPI-anchored [36–38]. Whereas H60a is widely expressed, H60b and c proteins display highly tissue- (but not mouse strain)-specific expression patterns [19]. Murine UL-16-binding protein-like transcript 1 (MULT1), which binds NKG2D with the highest affinity of all ligands, is so far the only transmembrane glycoprotein that belongs to the third group of murine NKG2D-binding proteins [39, 40].
In humans, NKG2D binds to the MHC class-I related proteins MICA and MICB (MHC class I chain-related protein A and B) [1]. NKG2D also recognizes surface glycoproteins that bind human cytomegalovirus (HCMV) UL-16 protein (ULBPs) [12, 41–43]. There are six members of the ULBP family of proteins, which are closely related to the Rae1 molecules in mice. ULBP1, -2, and -3 and -6 are GPI-anchored, while RAET1E and -G (also known as ULBP4 and ULBP5) are transmembrane proteins. NKG2D ligands are mostly induced by cellular stress (“induced self”), although some of them are expressed at low levels in different tissues [37–39, 44]. However, expression of the ligands is very tightly controlled at transcriptional or/and posttranscriptional levels, since unbalanced expression may trigger activation of the immune system and autoimmune responses [45].
NKG2D in the control of viral infections
Viruses, as intracellular pathogens, are usually efficiently controlled by NK and CD8+ T cells. NK cells limit viral replication and viral load in different tissues early upon infection, while CD8+ αβ T cells are responsible for the final viral clearance and, in some cases, for the establishment of viral latency. NKG2D is expressed on both of these cell types and plays an important role in the control of viral infections. Different viruses are able to induce expression of NKG2D ligands on infected cells [46–49]. They cause strong activation of NK cells as well as enhancement of antiviral effector functions of activated CD8+ αβ T cells, which can be prevented by NKG2D-specific mAbs [1, 13, 46, 48, 50]. Thus, NKG2D-mediated control represents a powerful and efficient mechanism to cope with viral infections.
However, the NKG2D-mediated immunosurveillance may exert considerable selective pressure on viruses in the course of the co-evolution with their hosts. Therefore, it is not surprising that some of them have developed mechanisms to evade NKG2D-mediated activation of the immune system [14].
Cytomegaloviruses (CMVs) have particularly well-developed mechanisms of interference with NKG2D-mediated immune cell activation. The HCMV-encoded membrane glycoprotein UL16 binds the NKG2D ligands ULBP1, ULBP2, and MICB [12, 51, 52], thus preventing their expression on the cell surface. HCMV gene product UL142 retains ULBP-3 in the cis-Golgi complex and prevents its transport to the membrane [53]. UL142 also retains newly synthesized full-length MICA (allele 001) in the cis-Golgi [54, 55]. Interestingly, the MICA allele 008, which lacks the cytoplasmatic domain, shows resistance to UL142 binding. The fact that the truncated form of MICA is the most common allele in several human populations suggests a profound advantage of the hosts’ ability to control virus infection through NKG2D-dependent activation of NK cells [56].
Although MICA and MICB genes are transcribed in all cells [57], their translation in healthy cells is repressed by endogenous cellular miRNAs [58]. Viruses exploit these structures to prevent NKG2D ligand upregulation. For example, the HCMV-derived miRNA, miR-UL112, protects infected cells from NKG2D-mediated lysis by acting as a sponge for MICB mRNA [59].
Mouse cytomegalovirus (MCMV) has also been shown to extensively downregulate NKG2D ligands. Four viral proteins are involved in this interference. MCMV protein m145 downmodulates cell surface expression of MULT-1 [60], m152 interferes with the expression of Rae1 and H60 proteins [61, 62], while m155 is involved in downmodulation of H60 [63]. In addition, m138/fcr-1 plays an important role in downmodulation of MULT-1, Rae1ε and H60 [64, 65].
Kaposi’s sarcoma-associated herpesvirus (KSHV) immune evasion gene K5 reduces cell surface expression of MICA and MICB as well as of the NKp80 ligand activation-induced C-type lectin (AICL) via ubiquitination of lysine residues in cytoplasmic tails of the ligands [66].
Epstein–Barr virus (EBV) can cause B cell lymphomas in immunosuppressed patients. EBV-transformed B cell lines express a high level of MHC class I molecules rendering them NK cell-resistant. However, reactivating latent EBV in transformed B cells increases susceptibility to NKG2D- and DNAM1-dependent NK cell lysis [67].
Ectromelia virus (ECTV) belongs to the orthopoxvirus genus, which includes variola virus (VARV), the causative agent of smallpox, and vaccinia virus (VV). Depletion of NK cells in mousepox-resistant mouse strains (C57BL/6) results in massive viremia and death [68]. ECTV induces expression of NKG2D ligands on infected cells. Neutralization of NKG2D recognition by antibodies causes increased viral titers and mortality [46]. Zoonotic orthopoxviruses (monkeypox and cowpox viruses) encode for a soluble antagonist of NKG2D [69]: orthopoxvirus MHC class I-like protein (OMCP). OMCP, which is conserved among cowpox and monkeypox viruses, is secreted by infected cells and blocks recognition of NKG2D ligands [69]. Binding of OMCP causes internalization of NKG2D, thus lowering the amount of available NKG2D receptor and inhibiting NKG2D-dependent killing by NK cells [69].
Human immunodeficiency virus (HIV)-encoded protein Nef (negative factor) has been shown to downregulate expression of the NKG2D ligands MICA, ULBP1, and ULBP2 on Jurkat and primary CD4+ T cells in vitro [70], in addition to HLA-A and -B [71]. In contrast to Nef, another HIV-encoded protein Vpr (viral protein R) has recently been shown to upregulate expression of NKG2D ligands ULBP1, -2, -3, but not MICA or MICB, both in vitro and in vivo [72]. Vpr causes upregulation of the ligands through activation of DNA damage/stress-sensing ATR kinase [48]. It seems that Vpr not only contributes to HIV-1-induced CD4+ T-lymphocyte depletion but also takes part in HIV-1-induced NK cell dysfunction [72].
In conclusion, NKG2D-mediated control appears to be an important and powerful mechanism in the immunosurveillance of viral infections. A number of viral evasion mechanisms targeting the NKG2D pathway just emphasize its importance and this fact should be seriously considered in the development of future vaccines.
NKG2D in the control of cancer
The potential role of NKG2D in cytotoxic anti-tumor responses was quickly recognized and discussed as a revival of the tumor surveillance model [73]. Early experiments showed that overexpression of NKG2D ligands in cancer cells caused tumor rejection after transplantation in mice [74, 75]. In humans, it was found that expression of NKG2D ligands highly correlated with the amount of T cell infiltrates in solid tumors [76]. Paradoxically, some tumors were shown to actively upregulate NKG2D ligands, making them prone to recognition by NK cells and cytotoxic T cells [76]. Various explanations have been proposed for this observation. As described in the previous section, viral infection leads to upregulation of NKG2D ligands. Some virally induced tumors therefore show enhanced expression of these proteins in vivo [77]. However, also tumors without viral origin can have high levels of NKG2D ligands, suggesting a different cause. One hypothesis is that induction of NKG2D ligands is the result of the oncogenic process itself. NKG2D ligands are stress-response genes and are upregulated by stimuli such as DNA damage [20, 78], heat-shock [44, 79], and shifts in hormone levels [80]. Many, if not all of these process readily occur in cancer cells. Oncogenesis is usually associated with genomic instability [81], heat-shock proteins are often hijacked by tumor cells to promote their survival [82] and many tumors, such as breast, endometrial and ovarian cancers use estrogens to promote their growth [83]. In addition, rapidly proliferating cells have been shown to induce NKG2D ligands [84]. Upregulation of these molecules in cancer cells might therefore be a bystander effect of the oncogenic process. Preventing lysis by NKG2D-expressing NK and CD8 T cells must therefore be a feature that a cancer cell acquires in order to sustain its growth.
Multiple strategies are used by cancer cells in order to prevent NKG2D mediated killing. The first and most obvious strategy is downmodulation of NKG2D ligands. Despite significant amounts of DNA damage or expression of heat-shock proteins, many tumors have been shown to actively inhibit NKG2D ligand expression [9, 85–87]. An important mechanism for NKG2D ligand downregulation appears to be the production of immunomodulatory cytokines such as TGFβ, which can be directly excreted by tumor cells themselves, or by regulatory immune cells that are expanded during tumor progression [88–91]. In addition to this system, tumors decrease surface NKG2D ligand expression via shedding of the extracellular domain by metalloproteases or with the assistance of the disulphide-isomerase ERp5 [92, 93]. Thirdly, expression of NKG2D ligands is actively regulated by microRNAs, and tumor cells also have been shown to manipulate this system via miRNA overexpression [58]. Experimental evidence for the in vivo relevance of NKG2D ligand downmodulation in the formation of tumors was recently shown in mice deficient for NKG2D. In animal models for spontaneous tumor formation, absence of NKG2D resulted in enhanced formation of aggressive tumors. In addition, these tumors were shown to have increased expression of Rae1 molecules on their surface [9].
Some tumors exploit an opposite strategy to manipulate NKG2D mediated signaling and instead induce high levels of its ligands [94]. This concept remained relatively ill-understood until it was mimicked in mice overexpressing Rae1 and MICA molecules [15, 95, 96]. In these animals, surface expression of NKG2D was greatly reduced, both on NK cells and on CD8 T cells, resulting in impaired anti-tumor responses by these immune cells [15, 95]. Interestingly, the impact of NKG2D ligand overexpression on CD8 T cell responses against pathogens appears to depend highly on the model used. Infection of transgenic mice with L. monocytogenes resulted in reduced anti-bacterial CD8 T cell numbers [15]. Antiviral responses upon mCMV infection, on the other hand, did not functionally impair CD8 T cell responses [96]. One explanation for these observations may be the differential co-stimulation of CD8 T cells upon infection with different pathogens. Unlike CD28 triggering, NKG2D co-stimulation is not required for T cell function [97]. Rather, NKG2D engagement appears to enhance the cytotoxic capacity of these cells. It will therefore depend on the pathogen or kind of tumor encountered to what extent NKG2D signaling is required for T cell-mediated cytotoxicity.
For NK cells, NKG2D is a directly activating receptor and NK cell function was impaired in MICA- and Rae1ε-transgenic animals. Not surprisingly, both in vivo and in vitro killing of tumor cells expressing NKG2D ligands by NK cells of these mice was reduced. Oppenheim and coworkers also suggested that constitutive engagement of NKG2D impaired NK cell function beyond its downmodulation and subsequent inability to engage its ligands on tumor cells [95]. However, other studies that directly addressed this issue indicate that this is not the case [15, 96]. Interestingly, NKG2D downmodulation via hyperstimulation appears to have different effects than inhibition of NKG2D signaling by omitting the molecules involved in transducing NKG2D-signaling. Downmodulation of NKG2D via antibody treatment or in Rae1 transgenic animals resulted in increased tumor cell growth in a model of chemically induced cancer formation [95, 98]. However, the same model showed no differences when NKG2D-deficient animals were compared with wild type controls [9]. The explanation for these differences is currently lacking, but it appears likely that NKG2D hyperligation induces compensatory and/or regulatory mechanisms which are absent in NKG2D-deficient animals.
In support of this notion is the observation that NKG2D stimulation promotes the specific outgrowth of regulatory T cell subsets in humans. NKG2D ligation enhances proliferation of a characteristic regulatory NKG2D+CD4+ T cell pool that is rare under normal conditions [6, 99]. This cell subset produces IL-10 and TGFβ, thus inhibiting immune responses in a paracrine fashion [99]. In addition, these cells express high levels of Fas ligand (FASL), which induces apoptosis in neighboring activated T cells, whereas these regulatory cells themselves appear refractory to this FASL [6]. Apart from directly presenting NKG2D ligands in cis, human tumor cells can manipulate the immune system by releasing a soluble form of NKG2D ligands. With the help of metalloproteases or of the cell surface-bound protein ERp5, the NKG2D ligands MICA, MICB, and ULBP2 are cleaved of the membrane and are capable of affecting NKG2D expressing cells in trans [100–102]. In addition, ULBP4 can be expressed in soluble form via alternative splicing [103]. Apart from producing soluble proteins, tumor cells produce exosomes with high levels of NKG2D ligands [104, 105]. Both soluble ligands and ligands expressed on exosomes have been shown to downmodulate NKG2D on cytotoxic cells and thus impair their anti-tumor activity.
In summary, it appears that tumor cells in general adapt methods to inhibit NKG2D signaling. They do this either via downmodulation of NKG2D ligands or via hyperexpression of NKG2D ligands on their surface or in soluble form. In addition, however, there are also tumors that do not seem to modify the NKG2D signaling pathway at all, yet still escape from destruction. These tumors also arise in experimental models [9], indicating that there is a ‘third’ method to avoid NKG2D-mediated killing. The molecular mechanism behind this phenomenon has yet to be revealed.
Due to its prominent role in tumor cell biology, the NKG2D signaling pathway has been under extensive investigation in the cancer field, both as a diagnostic tool and as a therapeutic target. MICA has been shown to be one of the most polymorphic genes within the group of MHC class I-related molecules [106] and several alleles, supposedly of reduced NKG2D affinity, have been associated with cancer [107–109]. Also, expression of NKG2D ligands, both the soluble and the membrane-bound form, has been shown to be a reliable marker for disease progression in a variety of malignancies [85, 94, 110], illustrating its value as a clinical marker.
Induction of NKG2D ligand expression on tumors appears to be a promising therapeutic strategy in cancer. Expression of the NKG2D ligand MULT1 is low under homeostatic conditions, due to its continuous ubiquitination and subsequent targeting to the proteasome. In stressed cells, this ubiquitination process is inhibited, leading to rapid upregulation of surface-bound MULT1 [45, 111]. In tumor cells, NKG2D ligand expression can therefore be specifically induced with proteasome inhibitors, making tumors sensitive to NKG2D-mediated killing [112]. In addition, HDAC inhibitors appear to stimulate NKG2D ligand expression on tumor cells and their immunogenicity for allogeneic NK cells [113].
Finally, the NKG2D receptor system has been used as a target for anti-cancer therapy. Therapeutic MICA-specific antibodies effectively opsonized cancer cells and induced DC-mediated cross-presentation of tumor antigens [114]. Bifunctional proteins consisting of a tumor-antigen directed antibody fused to NKG2D ligands effectively ‘coated’ tumor cells with activating ligands and increased their killing [115, 116].
In summary, the two sides of NKG2D in tumor biology make this protein a key molecule of interest for future oncological research. On one hand, the NKG2D receptor system may be exploited in anti-tumor strategies, and on the other hand, over-stimulation of NKG2D should be prevented in order to potentiate tumor surveillance.
NKG2D in autoimmunity
As previously mentioned, inappropriate or deregulated expression of NKG2D ligands on healthy cells can break the delicate balance between immune activation and tolerance, and trigger autoimmune response [117]. Several autoimmune diseases have therefore been associated with NKG2D signaling.
Insulin-dependent diabetes mellitus/type 1 diabetes (T1D) is a chronic autoimmune disorder in which insulin-producing Langerhans islets are destroyed by autoreactive immune cells. Genetic linkage studies have shown that some MICA alleles are positively associated with human T1D, but the functional relevance of this polymorphism is far from clear [118, 119]. The nonobese diabetic (NOD) mouse is widely studied as a model of human T1D [120]. Prediabetic NOD mice express Rae1 ligands on their islet cells [121]. The development of disease can be completely prevented by treatment with NKG2D-blocking mAbs, which reduce expansion and function of autoreactive CD8+ T cells [10].
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disorder in which immune cells, especially T cells, cause inflammation and destruction of the joints [122]. RA patients have high levels of IL-15 and TNF-α in the sera and inflamed joints [5], which induce expression of NKG2D on CD4+CD28− subset of T cells [5]. Since MICA and MICB molecules are also dramatically upregulated in RA synoviocytes, they activate the T cells in an NKG2D-dependent manner [5].
Celiac disease is an autoimmune disorder of the small intestine that occurs in genetically susceptible individuals as a reaction to wheat gliadin protein [123]. One of the diagnostic hallmarks of celiac disease is a massive infiltration of intraepithelial NKG2D+ CD8+ γδ T lymphocytes (IELs) in the gut [124]. MIC proteins, normally found intracellularly in enterocytes, become strongly expressed on the surface of epithelial cells in patients with active disease. There is evidence that this process is IL-15-dependent [125, 126].
Crohn’s disease (CD) belongs to the group of inflammatory bowel diseases. Significantly increased levels of MIC expression were found on intestinal epithelial cells. It has been shown that the number of intestinal epithelial CD4+ T cells expressing NKG2D highly correlates with the amount of intestinal inflammation and therefore are thought to play an important role in disease progression [4].
NKG2D in NK cell development
NKG2D is expressed very early in the development of NK cells, already at the stage of NK precursors (NKPs), the earliest NK committed cells [127]. About 10% of CD122+ NK1.1− NKPs (stage I) exhibit this receptor on the cell surface, whereas all immature NK cells (stage II and III) express NKG2D. During these stages of NK cell development, NKG2D expression rapidly increases and remains high through all later stages of maturation [127, 128].
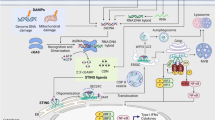
Recently, the intracellular signaling components of IL-15R and NKG2D have been shown to be coupled [129]. IL-15 is an essential cytokine for the development and survival of NK cells [130, 131]. Mice deficient for IL-15, IL-15Rβ, or any of the components of the IL-15 signaling pathway (e.g., Jak3, STAT5) have severe defects in NK cell development [132–136]. Horng et al. [129] have created a transgenic mouse strain expressing a DAP10 protein fused with the monoubiquitin (DAP10-Ub), which directly targets this molecule for proteasomal degradation. These mice display completely abolished NKG2D expression on T cells and severely reduced expression of this receptor on NK cells, which also express the NKG2D adaptor molecule DAP12. They observed severe defects in NK cell development because of their failure to respond to IL-15 stimulation. Jak3 has been shown to be the essential kinase downstream of the IL-15 receptor that is responsible for DAP10 phosphorylation important for the activation of STAT5, a prime target of Jak3. In addition, it was found that DAP10 can associate with the IL-15R β and γ (Fig. 1). Interestingly, in contrast to DAP10-Ub mice, defects in NK cell development were not reported in Dap10 −/− mice [23]. This could be explained by redundancy of the system, since intact NKG2D–DAP12 complexes on the surface of NK cells could compensate the lack of DAP10 to a major extent, despite a reduction in NKG2D expression. Activation of Syk by DAP12 can regulate survival and cytotoxicity via activation of PI3K [137, 138].

Coupling of the IL-15R and NKG2D signaling components in NK cells. The figure roughly summarizes interactions between signaling components of the IL-15R and NKG2D, which was proposed by Horng et al. [129]. Jak3, as a part of canonical IL-15R signaling pathway, is essential for the phosphorylation of DAP10. Activated DAP10 recruits PI3K and Grb2, which control proliferation, survival, and cytotoxicity of NK cells. DAP10 also associates with IL-15Rβ and -γ chains
Considering the above-mentioned findings, the question is whether and how NKG2D is involved in the development of NK cells. In transgenic mouse models with sustained expression of NKG2D ligands (MICA, Rae1ε, Rae1γ) and consequent downmodulation of NKG2D, impairments in the NK cell development were not observed [15, 95, 96]. In these models this may be the result of low-level expression of the receptor complexes on the cell surface and persistent NK cell stimulation.
To resolve this issue, recently two models of NKG2D-deficiency were reported [9, 139]. In the Klrk1 −/− mice generated by Guerra et al. [9], the authors did not find any major developmental defects, although they showed mild changes in some subpopulations of NK cells (i.e. Ly49A+ cells). In Klrk1 −/− mice generated in our lab, NK cell development was moderately affected [139]. In the absence of NKG2D we observed perturbations in the size of some developmental subsets of NK cells, increased proliferation of immature NK cells (mostly in stage II), their faster maturation and increased sensitivity to apoptosis. In addition, NK cell-mediated control of MCMV infection in these mice was better than in littermate controls, which we ascribed to the dysregulation and faster maturation of NK cells in the absence of NKG2D.
Differences in targeting strategies may be responsible for the observed effects. Our targeting inserted the EGFP sequence in the third exon of the Klrk1 locus, which could, as a foreign genetic element, cause non-specific effects. Therefore, we generated Klrk1 ∆/∆ mice in which the EGFP cassette is not present (Fig. 2a). These mice were obtained from the breeding of Klrk1 flox/flox mice (our unpublished data) and Cre ‘deleter’ mice, which resulted in the elimination of the floxed second and third exon of the Klrk1 gene in vivo, a mutation that was further propagated as Klrk1 ∆/∆. After we confirmed that NK cells from Klrk1 ∆/∆ mice do not express NKG2D (Fig. 2b), we analyzed the various stages of NK cell development in Klrk1 −/− versus Klrk1 ∆/∆ mice in comparison to wt animals (Fig. 3a). Klrk1 ∆/∆ mice displayed alterations in cell surface marker expression and maturation stages on NK cell subsets that were very similar to those seen in Klrk1 −/− animals. In addition, both mouse strains controlled early MCMV replication in a highly comparable manner (Fig. 3b). Thus, these data strongly argue that the previously observed phenotype of NK cells in our Klrk1 −/− mice [139] is the consequence of the desired Klrk1 mutation rather than of a genetic deregulation caused by the EGFP sequence.

Comparison of Klrk1 −/− and Klrk1 ∆/∆ mice. a Different mutations of the Klrk1 locus (upper row) in Klrk1 −/− (middle row) and Klrk1 ∆/∆ (lower row) mice are shown. Klrk1 ∆/∆ mice were obtained from the breeding of Klrk1 flox/flox and Cre deleter mice. b NKG2D expression on CD3−CD19−NK1.1+ cells isolated from the spleen of Klrk1 −/− (left panel) and Klrk1 ∆/∆ (right panel) mice is shown

Comparative analysis of NK cell phenotype and function between wild type, Klrk1 -/- and Klrk1 ∆/∆ mice. a NK cells isolated from the spleens of 9 weeks old Klrk1 −/− and Klrk1 ∆/∆ mice were analyzed by flow cytometry. Klrk +/+ littermates (B6) were used as controls. The data represent mean values of at least five mice per group. Error bars are indicated. b Indicated groups of Klrk1 −/−, Klrk1 ∆/∆ and their littermate controls were infected intravenously with 5 × 105 PFU of ∆m157 MCMV. The groups of mice were either NK cell depleted (PK136) or treated with PBS. Each symbol represents individual virus titers in indicated organs
In conclusion, NKG2D appears to play an important role in NK cell development. It seems that its role in the early development of NK cells is different than during the mature stages where NKG2D is responsible for activation of effector functions. “Unleashed” proliferation of immature NK cells and their increased sensitivity to apoptosis in the absence of NKG2D suggest that this receptor plays rather a regulatory role at this stage, most probably in concert with IL-15R and perhaps some other receptors (Fig. 1), controlling proliferation and survival of NK cells. Future research must reveal whether this function is limited to NK cells or whether other cellular subsets that constitutively express NKG2D (most notably NKT and γδ T cells) are also affected.
References
Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T (1999) Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285:727–729
Raulet DH (2003) Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 3:781–790
Saez-Borderias A, Guma M, Angulo A, Bellosillo B, Pende D, Lopez-Botet M (2006) Expression and function of NKG2D in CD4+ T cells specific for human cytomegalovirus. Eur J Immunol 36:3198–3206
Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, Caillat-Zucman S, Paul P, Gornet JM, Douay C, Ravet S, Tamouza R, Charron D, Lemann M, Mayer L, Toubert A (2007) CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology 132:2346–2358
Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T (2003) Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci USA 100:9452–9457
Groh V, Smythe K, Dai Z, Spies T (2006) Fas-ligand-mediated paracrine T cell regulation by the receptor NKG2D in tumor immunity. Nat Immunol 7:755–762
Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434:864–870
Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434:907–913
Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, Knoblaugh S, Cado D, Greenberg NM, Raulet DH (2008) NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28:571–580
Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, Lanier LL (2004) NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity 20:757–767
Ogasawara K, Benjamin J, Takaki R, Phillips JH, Lanier LL (2005) Function of NKG2D in natural killer cell-mediated rejection of mouse bone marrow grafts. Nat Immunol 6:938–945
Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ (2001) ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14:123–133
Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T (2001) Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol 2:255–260
Jonjic S, Babic M, Polic B, Krmpotic A (2008) Immune evasion of natural killer cells by viruses. Curr Opin Immunol 20:30–38
Wiemann K, Mittrucker HW, Feger U, Welte SA, Yokoyama WM, Spies T, Rammensee HG, Steinle A (2005) Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J Immunol 175:720–729
Nausch N, Cerwenka A (2008) NKG2D ligands in tumor immunity. Oncogene 27:5944–5958
Ho EL, Heusel JW, Brown MG, Matsumoto K, Scalzo AA, Yokoyama WM (1998) Murine NKG2D and Cd94 are clustered within the natural killer complex and are expressed independently in natural killer cells. Proc Natl Acad Sci USA 95:6320–6325
Glienke J, Sobanov Y, Brostjan C, Steffens C, Nguyen C, Lehrach H, Hofer E, Francis F (1998) The genomic organization of NKG2C, E, F, and D receptor genes in the human natural killer gene complex. Immunogenetics 48:163–173
Champsaur M, Lanier LL (2010) Effect of NKG2D ligand expression on host immune responses. Immunol Rev 235:267–285
Gasser S, Orsulic S, Brown EJ, Raulet DH (2005) The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436:1186–1190
Houchins JP, Yabe T, McSherry C, Bach FH (1991) DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J Exp Med 173:1017–1020
Diefenbach A, Tomasello E, Lucas M, Jamieson AM, Hsia JK, Vivier E, Raulet DH (2002) Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol 3:1142–1149
Gilfillan S, Ho EL, Cella M, Yokoyama WM, Colonna M (2002) NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat Immunol 3:1150–1155
Garrity D, Call ME, Feng J, Wucherpfennig KW (2005) The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc Natl Acad Sci USA 102:7641–7646
Rosen DB, Araki M, Hamerman JA, Chen T, Yamamura T, Lanier LL (2004) A Structural basis for the association of DAP12 with mouse, but not human, NKG2D. J Immunol 173:2470–2478
Chang C, Dietrich J, Harpur AG, Lindquist JA, Haude A, Loke YW, King A, Colonna M, Trowsdale J, Wilson MJ (1999) Cutting edge: KAP10, a novel transmembrane adapter protein genetically linked to DAP12 but with unique signaling properties. J Immunol 163:4651–4654
Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, Phillips JH (1999) An activating immunoreceptor complex formed by NKG2D and DAP10. Science (New York) 285:730–732
Wu J, Cherwinski H, Spies T, Phillips JH, Lanier LL (2000) DAP10 and DAP12 form distinct, but functionally cooperative, receptor complexes in natural killer cells. J Exp Med 192:1059–1068
Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH (1998) Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature 391:703–707
Karimi M, Cao TM, Baker JA, Verneris MR, Soares L, Negrin RS (2005) Silencing human NKG2D, DAP10, and DAP12 reduces cytotoxicity of activated CD8+ T cells and NK cells. J Immunol 175:7819–7828
McFarland BJ, Kortemme T, Yu SF, Baker D, Strong RK (2003) Symmetry recognizing asymmetry: analysis of the interactions between the C-type lectin-like immunoreceptor NKG2D and MHC class I-like ligands. Structure 11:411–422
McFarland BJ, Strong RK (2003) Thermodynamic analysis of degenerate recognition by the NKG2D immunoreceptor: not induced fit but rigid adaptation. Immunity 19:803–812
Wolan DW, Teyton L, Rudolph MG, Villmow B, Bauer S, Busch DH, Wilson IA (2001) Crystal structure of the murine NK cell-activating receptor NKG2D at 1.95 A. Nat Immunol 2:248–254
Cerwenka A, Bakker AB, McClanahan T, Wagner J, Wu J, Phillips JH, Lanier LL (2000) Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity 12:721–727
Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH (2000) Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol 1:119–126
Malarkannan S, Shih PP, Eden PA, Horng T, Zuberi AR, Christianson G, Roopenian D, Shastri N (1998) The molecular and functional characterization of a dominant minor H antigen, H60. J Immunol 161:3501–3509
Takada A, Yoshida S, Kajikawa M, Miyatake Y, Tomaru U, Sakai M, Chiba H, Maenaka K, Kohda D, Fugo K, Kasahara M (2008) Two novel NKG2D ligands of the mouse H60 family with differential expression patterns and binding affinities to NKG2D. J Immunol 180:1678–1685
Whang MI, Guerra N, Raulet DH (2009) Costimulation of dendritic epidermal gammadelta T cells by a new NKG2D ligand expressed specifically in the skin. J Immunol 182:4557–4564
Carayannopoulos LN, Naidenko OV, Fremont DH, Yokoyama WM (2002) Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J Immunol 169:4079–4083
Diefenbach A, Hsia JK, Hsiung MY, Raulet DH (2003) A novel ligand for the NKG2D receptor activates NK cells and macrophages and induces tumor immunity. Eur J Immunol 33:381–391
Bacon L, Eagle RA, Meyer M, Easom N, Young NT, Trowsdale J (2004) Two human ULBP/RAET1 molecules with transmembrane regions are ligands for NKG2D. J Immunol 173:1078–1084
Radosavljevic M, Cuillerier B, Wilson MJ, Clement O, Wicker S, Gilfillan S, Beck S, Trowsdale J, Bahram S (2002) A cluster of ten novel MHC class I related genes on human chromosome 6q24.2–q25.3. Genomics 79:114–123
Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, Spies T (2001) Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics 53:279–287
Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T (1996) Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci USA 93:12445–12450
Nice TJ, Coscoy L, Raulet DH (2009) Posttranslational regulation of the NKG2D ligand Mult1 in response to cell stress. J Exp Med 206:287–298
Fang M, Lanier LL, Sigal LJ (2008) A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS Pathog 4:e30
Walsh KB, Lanier LL, Lane TE (2008) NKG2D receptor signaling enhances cytolytic activity by virus-specific CD8+ T cells: evidence for a protective role in virus-induced encephalitis. J Virol 82:3031–3044
Ward J, Bonaparte M, Sacks J, Guterman J, Fogli M, Mavilio D, Barker E (2007) HIV modulates the expression of ligands important in triggering natural killer cell cytotoxic responses on infected primary T-cell blasts. Blood 110:1207–1214
Draghi M, Pashine A, Sanjanwala B, Gendzekhadze K, Cantoni C, Cosman D, Moretta A, Valiante NM, Parham P (2007) NKp46 and NKG2D recognition of infected dendritic cells is necessary for NK cell activation in the human response to influenza infection. J Immunol 178:2688–2698
Chen Y, Wei H, Sun R, Dong Z, Zhang J, Tian Z (2007) Increased susceptibility to liver injury in hepatitis B virus transgenic mice involves NKG2D-ligand interaction and natural killer cells. Hepatology 46:706–715
Dunn C, Chalupny NJ, Sutherland CL, Dosch S, Sivakumar PV, Johnson DC, Cosman D (2003) Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J Exp Med 197:1427–1439
Welte SA, Sinzger C, Lutz SZ, Singh-Jasuja H, Sampaio KL, Eknigk U, Rammensee HG, Steinle A (2003) Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur J Immunol 33:194–203
Bennett NJ, Ashiru O, Morgan FJ, Pang Y, Okecha G, Eagle RA, Trowsdale J, Sissons JG, Wills MR (2010) Intracellular sequestration of the NKG2D ligand ULBP3 by human cytomegalovirus. J Immunol 185:1093–1102
Chalupny NJ, Rein-Weston A, Dosch S, Cosman D (2006) Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem Biophys Res Commun 346:175–181
Ashiru O, Bennett NJ, Boyle LH, Thomas M, Trowsdale J, Wills MR (2009) NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J Virol 83:12345–12354
Zhang Y, Lazaro AM, Lavingia B, Stastny P (2001) Typing for all known MICA alleles by group-specific PCR and SSOP. Hum Immunol 62:620–631
Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T (2007) Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol 178:961–969
Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, Mandelboim M, Mandelboim O (2008) Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol 9:1065–1073
Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, Horwitz E, Prokocimer Z, Prichard M, Hahn G, Goldman-Wohl D, Greenfield C, Yagel S, Hengel H, Altuvia Y, Margalit H, Mandelboim O (2007) Host immune system gene targeting by a viral miRNA. Science 317:376–381
Krmpotic A, Hasan M, Loewendorf A, Saulig T, Halenius A, Lenac T, Polic B, Bubic I, Kriegeskorte A, Pernjak-Pugel E, Messerle M, Hengel H, Busch DH, Koszinowski UH, Jonjic S (2005) NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J Exp Med 201:211–220
Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL (2003) NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med 197:1245–1253
Krmpotic A, Busch DH, Bubic I, Gebhardt F, Hengel H, Hasan M, Scalzo AA, Koszinowski UH, Jonjic S (2002) MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat Immunol 3:529–535
Hasan M, Krmpotic A, Ruzsics Z, Bubic I, Lenac T, Halenius A, Loewendorf A, Messerle M, Hengel H, Jonjic S, Koszinowski UH (2005) Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J Virol 79:2920–2930
Lenac T, Budt M, Arapovic J, Hasan M, Zimmermann A, Simic H, Krmpotic A, Messerle M, Ruzsics Z, Koszinowski UH, Hengel H, Jonjic S (2006) The herpesviral Fc receptor fcr-1 down-regulates the NKG2D ligands MULT-1 and H60. J Exp Med 203:1843–1850
Arapovic J, Lenac Rovis T, Reddy AB, Krmpotic A, Jonjic S (2009) Promiscuity of MCMV immunoevasin of NKG2D: m138/fcr-1 down-modulates RAE-1epsilon in addition to MULT-1 and H60. Mol Immunol 47:114–122
Thomas M, Boname JM, Field S, Nejentsev S, Salio M, Cerundolo V, Wills M, Lehner PJ (2008) Down-regulation of NKG2D and NKp80 ligands by Kaposi’s sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc Natl Acad Sci USA 105:1656–1661
Pappworth IY, Wang EC, Rowe M (2007) The switch from latent to productive infection in Epstein–Barr virus-infected B cells is associated with sensitization to NK cell killing. J Virol 81:474–482
Jacoby RO, Bhatt PN, Brownstein DG (1989) Evidence that NK cells and interferon are required for genetic resistance to lethal infection with Ectromelia virus. Arch Virol 108:49–58
Campbell JA, Trossman DS, Yokoyama WM, Carayannopoulos LN (2007) Zoonotic orthopoxviruses encode a high-affinity antagonist of NKG2D. J Exp Med 204:1311–1317
Cerboni C, Neri F, Casartelli N, Zingoni A, Cosman D, Rossi P, Santoni A, Doria M (2007) Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J Gen Virol 88:242–250
Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D (1999) The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671
Richard J, Sindhu S, Pham TN, Belzile JP, Cohen EA (2010) HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood 115:1354–1363
Lanier LL (2001) A renaissance for the tumor immunosurveillance hypothesis. Nat Med 7:1178–1180
Cerwenka A, Baron JL, Lanier LL (2001) Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci USA 98:11521–11526
Diefenbach A, Jensen ER, Jamieson AM, Raulet DH (2001) Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 413:165–171
Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T (1999) Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci USA 96:6879–6884
Gourzi P, Leonova T, Papavasiliou FN (2006) A role for activation-induced cytidine deaminase in the host response against a transforming retrovirus. Immunity 24:779–786
Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C, Petrucci MT, Guarini A, Foa R, Santoni A (2009) ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 113:3503–3511
Groh V, Steinle A, Bauer S, Spies T (1998) Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science 279:1737–1740
Basu S, Pioli PA, Conejo-Garcia J, Wira CR, Sentman CL (2008) Estradiol regulates MICA expression in human endometrial cells. Clin Immunol 129:325–332
Negrini S, Gorgoulis VG, Halazonetis TD (2010) Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol 11:220–228
Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130:1005–1018
Persson I (2000) Estrogens in the causation of breast, endometrial and ovarian cancers—evidence and hypotheses from epidemiological findings. J Steroid Biochem Mol Biol 74:357–364
Molinero LL, Fuertes MB, Rabinovich GA, Fainboim L, Zwirner NW (2002) Activation-induced expression of MICA on T lymphocytes involves engagement of CD3 and CD28. J Leukoc Biol 71:791–797
McGilvray RW, Eagle RA, Watson NF, Al-Attar A, Ball G, Jafferji I, Trowsdale J, Durrant LG (2009) NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res 15:6993–7002
Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, Pende D, Steinle A, Ferrone S, Pistoia V (2004) Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia 6:558–568
Sers C, Kuner R, Falk CS, Lund P, Sueltmann H, Braun M, Buness A, Ruschhaupt M, Conrad J, Mang-Fatehi S, Stelniec I, Krapfenbauer U, Poustka A, Schafer R (2009) Down-regulation of HLA Class I and NKG2D ligands through a concerted action of MAPK and DNA methyltransferases in colorectal cancer cells. Int J Cancer 125:1626–1639
Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, Weller M, Friese MA (2006) TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain 129:2416–2425
Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, Puig PE, Novault S, Escudier B, Vivier E, Lecesne A, Robert C, Blay JY, Bernard J, Caillat-Zucman S, Freitas A, Tursz T, Wagner-Ballon O, Capron C, Vainchencker W, Martin F, Zitvogel L (2005) CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med 202:1075–1085
Li H, Han Y, Guo Q, Zhang M, Cao X (2009) Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol 182:240–249
Lee JC, Lee KM, Kim DW, Heo DS (2004) Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol 172:7335–7340
Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, Strong RK, Groh V, Spies T (2007) Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 447:482–486
Salih HR, Rammensee HG, Steinle A (2002) Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol 169:4098–4102
McGilvray RW, Eagle RA, Rolland P, Jafferji I, Trowsdale J, Durrant LG (2010) ULBP2 and RAET1E NKG2D ligands are independent predictors of poor prognosis in ovarian cancer patients. Int J Cancer 127:1412–1420
Oppenheim DE, Roberts SJ, Clarke SL, Filler R, Lewis JM, Tigelaar RE, Girardi M, Hayday AC (2005) Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat Immunol 6:928–937
Champsaur M, Beilke JN, Ogasawara K, Koszinowski UH, Jonjic S, Lanier LL (2010) Intact NKG2D-independent function of NK cells chronically stimulated with the NKG2D ligand Rae-1. J Immunol 185:157–165
Rajasekaran K, Xiong V, Fong L, Gorski J, Malarkannan S (2010) Functional dichotomy between NKG2D and CD28-mediated co-stimulation in human CD8+ T cells. PLoS One 5
Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y (2005) NKG2D function protects the host from tumor initiation. J Exp Med 202:583–588
Dai Z, Turtle CJ, Booth GC, Riddell SR, Gooley TA, Stevens AM, Spies T, Groh V (2009) Normally occurring NKG2D+CD4+ T cells are immunosuppressive and inversely correlated with disease activity in juvenile-onset lupus. J Exp Med 206:793–805
Groh V, Wu J, Yee C, Spies T (2002) Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419:734–738
Waldhauer I, Steinle A (2006) Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res 66:2520–2526
Boutet P, Aguera-Gonzalez S, Atkinson S, Pennington CJ, Edwards DR, Murphy G, Reyburn HT, Vales-Gomez M (2009) Cutting edge: the metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J Immunol 182:49–53
Cao W, Xi X, Hao Z, Li W, Kong Y, Cui L, Ma C, Ba D, He W (2007) RAET1E2, a soluble isoform of the UL16-binding protein RAET1E produced by tumor cells, inhibits NKG2D-mediated NK cytotoxicity. J Biol Chem 282:18922–18928
Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, Reyburn HT (2010) Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res 70:481–489
Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z (2008) Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol 180:7249–7258
Fodil N, Laloux L, Wanner V, Pellet P, Hauptmann G, Mizuki N, Inoko H, Spies T, Theodorou I, Bahram S (1996) Allelic repertoire of the human MHC class I MICA gene. Immunogenetics 44:351–357
Jumnainsong A, Romphruk AV, Jearanaikoon P, Klumkrathok K, Romphruk A, Luanrattanakorn S, Leelayuwat C (2007) Association of polymorphic extracellular domains of MICA with cervical cancer in northeastern Thai population. Tissue Antigens 69:326–333
Reinders J, Rozemuller EH, Otten HG, van der Veken LT, Slootweg PJ, Tilanus MG (2007) HLA and MICA associations with head and neck squamous cell carcinoma. Oral Oncol 43:232–240
Tamaki S, Sanefuzi N, Ohgi K, Imai Y, Kawakami M, Yamamoto K, Ishitani A, Hatake K, Kirita T (2007) An association between the MICA-A5.1 allele and an increased susceptibility to oral squamous cell carcinoma in Japanese patients. J Oral Pathol Med 36:351–356
Nuckel H, Switala M, Sellmann L, Horn PA, Durig J, Duhrsen U, Kuppers R, Grosse-Wilde H, Rebmann V (2010) The prognostic significance of soluble NKG2D ligands in B-cell chronic lymphocytic leukemia. Leukemia 24:1152–1159
Nice TJ, Deng W, Coscoy L, Raulet DH (2010) Stress-regulated targeting of the NKG2D ligand Mult1 by a membrane-associated RING-CH family E3 ligase. J Immunol 185:5369–5376
Vales-Gomez M, Chisholm SE, Cassady-Cain RL, Roda-Navarro P, Reyburn HT (2008) Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res 68:1546–1554
Diermayr S, Himmelreich H, Durovic B, Mathys-Schneeberger A, Siegler U, Langenkamp U, Hofsteenge J, Gratwohl A, Tichelli A, Paluszewska M, Wiktor-Jedrzejczak W, Kalberer CP, Wodnar-Filipowicz A (2008) NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood 111:1428–1436
Jinushi M, Hodi FS, Dranoff G (2006) Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity. Proc Natl Acad Sci USA 103:9190–9195
Germain C, Campigna E, Salhi I, Morisseau S, Navarro-Teulon I, Mach JP, Pelegrin A, Robert B (2008) Redirecting NK cells mediated tumor cell lysis by a new recombinant bifunctional protein. Protein Eng Des Sel 21:665–672
von Strandmann EP, Hansen HP, Reiners KS, Schnell R, Borchmann P, Merkert S, Simhadri VR, Draube A, Reiser M, Purr I, Hallek M, Engert A (2006) A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood 107:1955–1962
Caillat-Zucman S (2006) How NKG2D ligands trigger autoimmunity? Hum Immunol 67:204–207
Nikitina-Zake L, Rajalingham R, Rumba I, Sanjeevi CB (2004) Killer cell immunoglobulin-like receptor genes in Latvian patients with type 1 diabetes mellitus and healthy controls. Ann N Y Acad Sci 1037:161–169
Bilbao JR, Martin-Pagola A, Vitoria JC, Zubillaga P, Ortiz L, Castano L (2002) HLA-DRB1 and MHC class 1 chain-related A haplotypes in Basque families with celiac disease. Tissue Antigens 60:71–76
Delovitch TL, Singh B (1997) The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity 7:727–738
Ogasawara K, Hamerman JA, Hsin H, Chikuma S, Bour-Jordan H, Chen T, Pertel T, Carnaud C, Bluestone JA, Lanier LL (2003) Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity 18:41–51
Van Belle TL, von Herrath MG (2009) The role of the activating receptor NKG2D in autoimmunity. Mol Immunol 47:8–11
Stepniak D, Koning F (2006) Celiac disease—sandwiched between innate and adaptive immunity. Hum Immunol 67:460–468
Bhagat G, Naiyer AJ, Shah JG, Harper J, Jabri B, Wang TC, Green PH, Manavalan JS (2008) Small intestinal CD8+TCRgammadelta+NKG2A+ intraepithelial lymphocytes have attributes of regulatory cells in patients with celiac disease. J Clin Invest 118:281–293
Mention JJ, Ben Ahmed M, Begue B, Barbe U, Verkarre V, Asnafi V, Colombel JF, Cugnenc PH, Ruemmele FM, McIntyre E, Brousse N, Cellier C, Cerf-Bensussan N (2003) Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology 125:730–745
Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, Raulet DH, Lanier LL, Groh V, Spies T, Ebert EC, Green PH, Jabri B (2004) Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 21:357–366
Huntington ND, Vosshenrich CA, Di Santo JP (2007) Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev 7:703–714
Yokoyama WM, Kim S, French AR (2004) The dynamic life of natural killer cells. Annu Rev Immunol 22:405–429
Horng T, Bezbradica JS, Medzhitov R (2007) NKG2D signaling is coupled to the interleukin 15 receptor signaling pathway. Nat Immunol 8:1345–1352
DiSanto JP, Muller W, Guy-Grand D, Fischer A, Rajewsky K (1995) Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci USA 92:377–381
Huntington ND, Puthalakath H, Gunn P, Naik E, Michalak EM, Smyth MJ, Tabarias H, Degli-Esposti MA, Dewson G, Willis SN, Motoyama N, Huang DC, Nutt SL, Tarlinton DM, Strasser A (2007) Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat Immunol 8:856–863
Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, Matsuki N, Charrier K, Sedger L, Willis CR, Brasel K, Morrissey PJ, Stocking K, Schuh JC, Joyce S, Peschon JJ (2000) Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med 191:771–780
Di Santo JP (2006) Natural killer cell developmental pathways: a question of balance. Annu Rev Immunol 24:257–286
Suzuki H, Duncan GS, Takimoto H, Mak TW (1997) Abnormal development of intestinal intraepithelial lymphocytes and peripheral natural killer cells in mice lacking the IL-2 receptor beta chain. J Exp Med 185:499–505
Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, Ma A (1998) IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity 9:669–676
Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, Doherty PC, Grosveld GC, Ihle JN (1995) Defective lymphoid development in mice lacking Jak3. Science 270:800–802
Jiang K, Zhong B, Gilvary DL, Corliss BC, Vivier E, Hong-Geller E, Wei S, Djeu JY (2002) Syk regulation of phosphoinositide 3-kinase-dependent NK cell function. J Immunol 168:3155–3164
Jiang K, Zhong B, Ritchey C, Gilvary DL, Hong-Geller E, Wei S, Djeu JY (2003) Regulation of Akt-dependent cell survival by Syk and Rac. Blood 101:236–244
Zafirova B, Mandaric S, Antulov R, Krmpotic A, Jonsson H, Yokoyama WM, Jonjic S, Polic B (2009) Altered NK cell development and enhanced NK cell-mediated resistance to mouse cytomegalovirus in NKG2D-deficient mice. Immunity 31:270–282
Acknowledgments
We would like to thank to Stipan Jonjić and Astrid Krmpotić for helpful discussions. We are very grateful to Sali Slavić Stupac and Miro Samsa for excellent technical assistance and animal care. This work was supported by Croatian Ministry of Science, Education and Sports (Grant No. 062-0621261-127), EU FP7-Regpot-2010-5 Grant TransMedRi (No. 256686) and Croatian-Israeli grant to B.P.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Zafirova, B., Wensveen, F.M., Gulin, M. et al. Regulation of immune cell function and differentiation by the NKG2D receptor. Cell. Mol. Life Sci. 68, 3519–3529 (2011). https://doi.org/10.1007/s00018-011-0797-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0797-0