
Abstract
Background
There are many studies analysing the effect of SNPs in genes coding proteins which are involved in innate immune response on susceptibility to invasive bacterial disease. Many of them gave inconclusive results. Regarding the complexity of immune response and cooperation between particular elements, number of SNPs may have a cumulative effect on the susceptibility to bacterial meningitis.
Findings
In most studies cooccurrence of several SNPs was not analysed. These studies were performed on small groups of patients and usually only few SNPs were checked simultaneously. Additionally, comparison of the results across the studies is hard to conduct. We hypothesise that the number of variants of genes involved in innate immune response plays a role in susceptibility to bacterial meningitis. However, the role of toll-like receptors and other part of innate immune response in the eradication of bacteria, and initiation of the inflammatory response in CNS need further studies.
Conclusion
Large multicentre studies assessing multiple SNPs in patients with microbiologically proven pneumococcal or meningococcal meningitis are needed to find real genetic risk factors for developing bacterial meningitis. This is necessary to design more effective treatment and prevention strategies for severe infections.
Similar content being viewed by others


Introduction
Bacterial meningitis (BM) is one of the most devastating infections in childhood with a high rate of mortality. Despite the common use of vaccination, the leading causes of bacterial meningitis are Streptococcus pneumoniae and Neisseria meningitidis [1,2,3]. Carriage of these bacteria is prevalent in general population. The question is why some people are only asymptomatic carriers while others develop the devastating disease. Most patients with BM were previously healthy, not suffering from chronic diseases, and not diagnosed with immunodeficiency. There are several steps to develop bacterial meningitis [4]. The first one is the colonisation of mucous membranes. Then, after invasion through mucous membranes bacteria reach the blood. Even with the protective blood–brain barrier, pathogens can reach the central nervous system (CNS). The final step is an inflammatory process in the cerebrospinal fluid.
Immune response in CNS is unique. In cerebrospinal fluid (CSF) antigen presenting cells (APC) are not present and the subarachnoid cavity has no lymphatic drainage [5]. The goal of innate immunity in CSF is the recognition and response to a pathogen. It is different from peripheral innate response which cooperates with adaptive immune response in spleen and lymph nodes [6, 7]. In most cases pathogens enter CNS via blood. They were previously presented to peripheral immune cells, which are responsible for generating the adaptive immune response.
Immune response in CNS must be well balanced, and absence of adaptive immunity in CNS allows to protect neuronal tissue from damage caused by inflammatory processes. The inflammatory response in CSF is stimulated by pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Among pattern recognition receptors (PRR) localised in leptomeninges and choroid plexus, toll-like receptors (TLR) are of crucial importance in antibacterial response [8,9,10].
Toll-like receptor was originally identified as a receptor essential for dorsoventral polarity in the early development of Drosophila [11]. TLRs are transmembrane PRRs; they constitute a large superfamily with interleukin 1 receptor (IL-1R) family. They are either expressed at the cell surface for extracellular ligand recognition or localised in endosomal compartments for the recognition of pathogen-associated nucleic acids. Among the characterised TLRs, TLR1, 2, 4, 5 and 6 are represented on the cell surface and recognise bacterial and fungal products, whereas TLR 3,7, 8 and 9 reside in the intracellular endosomes and specialise in the detection of pathogens’ nucleic acids [9, 12, 13]. Microglial cells express all TLRs identified to date; astrocytes express TLR 2, 3, and 9, neurons express TLR 3, 7, 8, and 9, and oligodendrocytes express TLR 2 and 3 [8]. The cytoplasmatic part of TLR possesses the domain with a high degree of similarity with IL-1R [9, 12]. Their horseshoe-like shape forms hetero or homodimers (M-shaped structure) to bind PAMPs. Signal transduction via TLR engages proteins such as myeloid differentiation factor 88 (MyD88), kinase IRAK (IL-interleukin-1R-associated protein kinases), kinase TAK1 (TGF-beta-activated kinase), kinase-binding protein TAK1, and TNF-receptor-associated factor 6 [14]. Within 4 h after TLR activation APCs, macrophages and natural killers (NK) are activated and production of cytokines (IL-1, IL-6, IL-12), TNF-alpha (TNF-α), interferon alpha/beta (IFN-α/β) occurs [14]. This activation cascade leads to pathogen elimination and inhibition of the inflammatory process through IL10 [14, 15]. Cytokines decrease the integrity of blood–brain barrier and increase leukocyte migration [6].
First data about TLR in pneumococcal meningitis were presented in paper by Echchannaoui et al. [16]. The authors shown more invasive diseases in TRL2-deficient mice. Nowadays, it is proven that three from ten different TLRs (TLR2, TLR4, and TLR9) are involved in inflammatory response to bacteria such as S. pneumoniae or N. meningitidis [17, 18].
The human TLR2 is stimulated by bacterial cell-wall components [20]. In a mouse model deficiency of TLR2 increases the severity of the pneumococcal meningitis [19]. TLR4 is found to be essential for lipopolysaccharide (LPS) signalling, but alone is not sufficient for LPS recognition. LPS is recognised by heterodimer of TLR4 and MD-2. A mutant of N. meningitidis which is LPS-lacking is not able to cause TLR4 dependent response [17].TLR4 is also stimulated by pneumococcal pneumolysin. TLR9 is an intracellular PRR which recognises phagocytosed unmethylated Cytosine–phosphate–Guanine (CpG) motives in bacterial DNA [9]. This protein is present in phagocytosing microglia and antigen presenting astrocytes in CSN. TLR9 activation triggers the MyD88-dependent pro-inflammatory pathway [19].
A recognition of certain PAMPs involves also a TLR-independent path. Of the best-characterised intracellular PRRs are the nucleotide-binding domain (NOD) proteins that recognise bacterial peptidoglycan. NOD-like receptors (NLRs) constitute a family of cytoplasmic PRRs, which contains more than 22 members [20,21,22]. NOD1 and NOD2 recognise peptidoglycans: NOD1 detects meso-DAP Gram negative and NOD2 recognises muramyl dipeptide, present in both Gram-positive and Gram-negative bacteria. NOD2 can also recognise single-strand RNA.
NLRs are characterised by a tripartite-domain organisation with a conserved NOD domain and leucine-rich repeats [23]. Their primary role is to recognise PAMPs and DAMPs to induce inflammasome formation and production of IL-1 and IL-18 via Caspase-1 activation. NLR binds to RIPK2 (receptor-interacting serine-threonine protein kinase 2) leading to nuclear factor-κB (NF-κB) and MAP kinase activation [23]. NOD-receptors are also involved in MyD88-dependent signalling cascade downstream of TLR2 and TLR4. Pneumococci can induce NF-κB activation through NOD2- and RIP2-dependent signalling [22].
Activation of NF-κB through a TLR or NOD-like receptor binding is considered to be the central regulator of the innate immunity. This mechanism is controlled by inhibitors which are encoded by genes NFKBIA, NFKBIE, and NFKBIZ (Nuclear Factor of Kappa Light Chain Gene Enhancer in B cells inhibitor) [24]. The degradation of NF-κB inhibitors, including IκB-α, IκB-ε, and IκB-ζ (encoded by the genes NFKBIA, NFKBIE, and NFKBIZ), leads to NF-κB translocation to the nucleus and gene transcription resulting in cytokine production [24].
TIRAP (toll–interleukin 1 receptor—TIR domain-containing adaptor protein) is an adaptor protein which is essential for the inflammatory response. TIRAP is involved in TLR2 and TLR4 signalling pathways leading to the activation of NF-κB, MAPK1, MAPK3 and JNK, and resulting in cytokine secretion and the inflammatory response. TIRAP also positively regulates the production of TNF-alpha and IL-6 [25].
There are no doubts that innate immune response is crucial in susceptibility to bacterial meningitis. Four Mendelian primary immunodeficiencies associated with impaired signalling of the TLR pathway have been reported with mutations in MyD88, interleukin-1 receptor-associated kinase-4 (IRAK4), nuclear factor-kappa B essential modulator (NEMO) and IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha) [26]. These diseases are caused by monogenic mutations [26]. The first described deficiency in TLR signalling is IRAK4 deficiency. This deficiency causes no response to TLR ligands including LPS. Cells after stimulation do not produce TNF-alpha, IL-1 beta, IL-12 p40, or IL-6 which results in recurrent staphylococcal and streptococcal infections [26, 27].
Mutations in genes coding proteins involved in innate immunity, although rare in general population, cause severe immunodeficiency manifesting with severe diseases in early childhood. On the other hand, the effect of single nucleotide polymorphisms (SNPs) in above-mentioned genes is not that harmful to a particular patient, but because SNPs are common—their global effect may be even more important.
Having in mind the complexity of the immune response, we see the need for analysis of SNPs in genes involved in naive immune response on bacterial meningitis. In this manuscript, we summarise the results from different studies to find out an association between SNPs in genes coding PRPs and susceptibility to bacterial meningitis (Table 1).
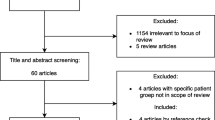
A review of the medical literature from the last 16 years (2001–2016) was performed in the primary database PubMed, using MeSH descriptors and combinations of the descriptors “SNPs”, “bacterial meningitis”, “pneumococcal meningitis”, “meningococcal meningitis”. Excluded from the review were studies which included newborns, particularly due to the different meningitis-causing agents in this age group.
TLR 2 and TLR4
There are two SNPs in TLR2 widely described in the literature: rs4696480 and rs5743708. TLR2 rs57437708 located in the Toll/IL-1 receptor region of TLR2 has an essential role in the formation of dimers [28]. The presence of this SNP induces a defective TLR2 dimerisation, and thus reduced response against meningococcal porin B [28, 29].
In the case of human TLR4, two common polymorphisms are found, rs4986790 and rs4986791, which are related to the hyporesponsiveness to LPS. Polymorphic variants are present in approximately 10% of Caucasian individuals [30]. As expected, people with these mutations are more frequently infected with Gram-negative bacteria than patients without mutations, and their condition is more severe. Although mutations are localised outside of the ligand-binding domain, they cause local conformational changes around the LPS-binding site, what results in decreased functional TLR4 expression. That change interferes with TLR4 interaction with MyD88. TLR4 rs4986790 mutated variant due to hyporesponsiveness to LPS may cause susceptibility to invasive meningococcal and pneumococcal infections [31].
Yuan et al. found a lower incidence of the TLR4 rs4986790 polymorphism in a group of 85 Australian patients with invasive pneumococcal disease [29]. TLR2 rs5743708 polymorphism was present in 4% children with pneumococcal meningitis and 7% of the control group (p = 0.04). This was a relatively small group of patients with invasive pneumococcal disease—not only meningitis. On the other hand, Moens et al. did not find any association between TLR2 rs4696480, TLR2 rs5743708, and TLR4 rs4986790 SNPs and susceptibility to invasive pneumococcal infection in 99 Belgian patients [28].
Statistically significant differences in TLR2 and TLR4 SNP frequency were found by Telleria-Orriols [31]. In the study group, there were 59 children with meningococcal infection (18 with meningococcal meningitis) and 114 with pneumococcal infection (only 12 with meningitis) of Caucasian origin. 59% of patients with meningitis carried at least one copy of a risk allele. In the control group, the risk allele was found in 16.6% children (p < 0.0001). This study showed two times higher frequency of TLR4 polymorphic variant in patients than in controls (23.7% vs 9.1%; p = 0.0472) [31].
Smirnova et al. analysed total nucleotide variation at TLR4 and TLR2 loci. The study group included 197 British patients with invasive meningococcal disease and only half of these patients had meningitis. Rare TLR4 coding variants were markedly over represented in patients compared to controls. Similar situation was observed with rare variants of TLR2 rs57437708, but this finding was not of statistical value [32].
In a study by Read et al. on 1047 British patients with proven meningococcal disease and 879 healthy controls, polymorphic variants of TLR4 were detected in 6.5% of patients and 5.9% of controls [33]. The difference was not of statistical significance. Biebl et al. checked SNPs in TLR4 in a group of Austrian survivors from meningococcal disease [34]. There were 185 patients with proven meningococcal infection and 770 healthy controls. Only 53% of patients had meningitis. Age of patients was widely scattered, ranging from 1 month to 74 years. TLR4 rs4986790 was not significantly associated with the susceptibility to invasive meningococcal disease [34].
Age of studied population appeared to be important factor. Faber et al. in a group of children younger than 12 months found 1.55 times increased risk of meningococcal meningitis for TLR4 rs4986790 variant carriers compared to controls [30]. Allen et al. in the study on meningitis did not show any association between TLR4 rs4986790 and TLR4 rs4986791 SNPs and susceptibility to meningococcal meningitis during epidemics in Gambian children [35].
Mutant alleles of NOD2 were associated with decreased activation of NF-κB [22]. In vitro studies showed that NOD2 is upregulated after exposure to N. meningitidis. Van Well et al. in the group of 391 children with meningococcal and 82 pneumococcal meningitis and 1141 controls showed that variants of TLR4 rs4986790 and NOD2 rs2066847 are significantly associated with meningococcal meningitis [36]. Polymorphic variants of NOD2 rs2066847 were more common in patients with meningococcal meningitis but not with pneumococcal meningitis. No differences were observed for TLR2 rs5743708.
TLR9
TLR9 rs352140 polymorphic allele creates an increased affinity for NF-κB which in turn increases the transcriptional activity of the gene leading to enhanced production of cytokines, what results in immune-mediated cochlear damage [37]. With respect to SNPs of TLR9 (rs352139 and rs352140), no differences in frequencies of genotypes or alleles were observed among patients with bacterial meningitis and controls in a study by Zhang on Chinese children [38]. They could not find any significant differences when combined analysis of the two SNPs of TLR9 was performed. Sanders et al. in a study on 390 paediatric survivors of meningococcal meningitis demonstrated a protective effect of TLR9 rs352140 variants [38].
TIRAP
Homozygous variants of TIRAP rs8177374 (which encode a leucine substitution at Ser180 of TIRAP) are rare in developing countries. It supports speculations that this homologous TIRAP rs8177374 variant may increase susceptibility to specific infectious diseases to such an extent that it may have selected itself out of population [25]. A recently discovered SNP in TIRAP rs8177374 impairs TLR2-mediated NF-κB signalling in reconstitution experiments [25]. The variant was less able to bind TLR2 in comparison with the wild one. Khor et al. showed a protective effect of heterozygous variant of TIRAP rs8177374 against pneumococcal infection, malaria, and tuberculosis [25].
NFKBIA, NFKBIE and NFKBIZ
In the study by Lundbo et al. on a large group of Danish children with pneumococcal or meningococcal meningitis, the authors have shown the association between NFKBIE SNP and susceptibility to pneumococcal meningitis [39]. They checked seven SNPs also in other inhibitory factors (NFKBIA, NFKBIL2, NFKBIZ, TIRAP), but no association was found.
In the study group of Chapman et al., there were 275 British patients with 733 controls, and 173 African patients with 550 controls [40]. It was found that common NFKBIL2 polymorphisms were associated with susceptibility to invasive pneumococcal disease in European and African populations. The NFKBIE SNP was associated with increased risk of pneumococcal meningitis, but not bacteraemia. The remaining SNPs were not associated with susceptibility to invasive disease. None of the SNPs were associated with risk of invasive meningococcal disease or mortality [39, 40].
Altogether, we were able to summarise the literature on SNPs that affects the susceptibility to pneumococcal and meningococcal meningitis. There are many studies analysing the effect of SNPs in genes coding proteins which are involved in innate immune response on susceptibility to invasive bacterial disease (including bacterial meningitis). Many of them gave inconclusive results. These studies were performed on small groups of patients and usually only few SNPs were checked simultaneously. Regarding the complexity of immune response and cooperation between particular elements, number of SNPs may have a cumulative effect on the susceptibility to bacterial meningitis. In most studies cooccurrence of several SNPs was not analysed.
Additionally, comparison of the results across the studies is hard to conduct. The studies were performed on different groups of patients: some were conducted on patients with meningitis, and some on patients with sepsis. In cohort studies the percentage of patients with meningitis of proved bacterial aetiology was estimated at 10%. The diagnosis of meningitis is clear, but in some studies patients with bacteraemia or sepsis were also included. The gold standard for establishing the aetiology is the microbiological culture. Molecular detection of pathogen genetic material is also accepted, but in some studies meningococcal aetiology was based on the clinical picture—sepsis with severe purpura. Control groups were also different. Utilising the blood donors as the controls gives an opportunity to collect big group, but this method poses many disadvantages. In such a control group, the information about diseases in childhood is lacking. Most studies collected data from meningitis survivors, thus patients with the severest disease are excluded. Many studies were based on a genetic material from banks, thus researchers did not have detailed clinical data on past medical history of those children.
A large number of studies have been published to date, but those studies involved relatively small groups of patients. Many of the associations have not been reproduced in larger studies, so future studies should involve large group of patients with well-defined conditions. Large multicentre studies assessing multiple SNPs in patients with microbiologically proven pneumococcal or meningococcal meningitis are needed to find real genetic risk factors for developing bacterial meningitis, what is necessary to design more effective treatment and prevention strategies for severe infections.
Abbreviations
- APC:
-
Antigen presenting cells
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DAMPS:
-
Damage-associated molecular patterns
- IκBα:
-
Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- IL:
-
Interleukin
- IFN-α/β:
-
Interferon alpha/beta
- IRAK4:
-
Interleukin-1 receptor-associated kinase-4
- LPS:
-
Lipopolysaccharide
- MyD:
-
Myeloid differentiation factor
- NEMO:
-
Nuclear factor-kappa B essential modulator
- NOD:
-
Nucleotide-binding oligomerization domain
- NLR:
-
NOD-like receptor
- NK:
-
Natural killers
- PAMP:
-
Pathogen-associated molecular pattern
- PGN:
-
Peptidoglycan
- PRR:
-
Pattern recognition receptor
- RIPK2:
-
Receptor-interacting serine-threonine protein kinase 2
- TLR:
-
Toll-like receptor
- TNF:
-
Tumour necrosis factor
- TAK1:
-
TGF-beta-activated kinase
- PID:
-
Primary immunodeficiencies
References
World Health Organization. Surveillance for vaccine preventable diseases (VPDs). http://www.who.int/immunization/monitoring_surveillance/ burden/VPDs/en/. Accessed Aug 2017.
CDC. Active bacterial core surveillance report, emerging infections program network, Neisseria meningitidis. https://www.cdc.gov/abcs/reports-findings/survreports/mening14.pdf. Accessed Aug 2017.
Borrow R, Alarcon P, Carlos J, Caugant DA, Christensen H, Debbag R, et al. The Global meningococcal initiative: global epidemiology, the impact of vaccines on meningococcal disease and the importance of herd protection. Expert Rev Vaccines. 2016;1–16.
Brouwer MC, Tunkel AR, van de Beek D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev. 2010;23:24.
Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J. 2009;420:1–16.
Pachter JS, De Vries HE, Fabry Z. The blood–brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol. 2003;62:593–604.
Johnston M, Zakharov A, Papaiconomou C, Salmasi G, Armstrong D. Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non-human primates and other mammalian species. Cerebrospinal Fluid Res. 2004;1:2.
Koedel U. Toll-like receptors in bacterial meningitis. Curr Top Microbiol Immunol. 2009;336:15–40.
Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511.
Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–21.
Medzhitov R, Preston-Hurlburt P, Janeway. CAJ.A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7.
Koedel U, Angele B, Rupprecht T, Wagner H, Roggenkamp A, Pfister HW, Kirschning CJ. Toll-Like receptor 2 participates in mediation of immune response in experimental pneumococcal meningitis. J Immunol. 2003;170:438–44.
Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–63.
Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell. 2006;126:659–62.
Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–89.
Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R. Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis. 2002;186:798–806.
Klein M, Angele B, Pfister HW, Wagner H, Koedel U, Kirschning CJ. Detection of pneumococcal infection of the central nervous system depends upon TLR2 and TLR4. J Infect Dis. 2008;198:1028–36.
Zughaier SM, Tzeng YL, Zimmer SM, Datta A, Carlson RW, Stephens DS. Neisseria meningitidis lipooligosaccharide structure-dependent activation of the macrophage CD14/Toll-like receptor 4 pathway. Infect Immun. 2004;72:371–80.
Mogensen TH, Paludan SR, Kilian M, Ostergaard L. Live Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis activate the inflammatory response through Toll-like receptors 2, 4, and 9 in species-specific patterns. J Leukoc Biol. 2006;80:267–77.
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801.
Kim YG, Park JH, Shaw MH, Franchi L, Inohara N, Nunez G. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to toll-like receptor ligands. Immunity. 2008;28:246–57.
Hasegawa M, Yang K, Hashimoto M, Park JH, Kim YG, Fujimoto Y, Nunez G, Fukase K, Inohara N. Differential release and distribution of Nod1 and Nod2 immunostimulatory molecules among bacterial species and environments. J Biol Chem. 2006;281:29054–63.
Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–82.
Chapman SJ, Khor CC, Vannberg FO, Rautanen A, Walley A, Segal S, Moore CE, Davies RJ, Day NP, Peshu N, Crook DW, Berkley JA, Williams TN, Scott JA, Hill AV. Common NFKBIL2 polymorphisms and susceptibility to pneumococcal disease: a genetic association study. Crit Care. 2010;14:R227.
Khor CC, Chapman SJ, Vannberg FO, Dunne A, Murphy C, Ling EY, Frodsham AJ, Walley AJ, Kyrieleis O, Khan A, Aucan C, Segal S, Moore CE, Knox K, Campbell SJ, Lienhardt C, Scott A, Aaby P, Sow OY, Grignani RT, Sillah J, Sirugo G, Peshu N, Williams TN, Maitland K, Davies RJ, Kwiatkowski DP, Day NP, Yala D, Crook DW, Marsh K, Berkley JA, O’Neill LA, Hill AV. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet. 2007;39:523–8.
Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais CM, Dupuis S, Feinberg J, Fieschi C, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–9. https://doi.org/10.1126/science.108190.
Medvedev AE, Lentschat A, Kuhns DB, Blanco JC, Salkowski C, Zhang S, Arditi M, Gallin JI, Vogel SN. Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. J Exp Med. 2003;198:521–31.
Moens L, Verhaegen J, Pierik M, Vermeire S, De BK, Peetermans WE, Bossuyt X. Toll-like receptor 2 and toll-like receptor 4 polymorphisms in invasive pneumococcal disease. Microbes Infect. 2007;9:15–20.
Yuan FF, Marks K, Wong M, Watson S, de Leon E, et al. Clinical relevance of TLR2, TLR4, CD14 and FcgammaRIIA gene polymorphisms in Streptococcus pneumoniae infection. Immunol Cell Biol. 2008;86:268–70.
Faber J, Meyer CU, Gemmer C, Russo A, Finn A, Murdoch C, Zenz W, Mannhalter C, Zabel BU, Schmitt HJ, Habermehl P, Zepp F, Knuf M. Human toll-like receptor 4 mutations are associated with susceptibility to invasive meningococcal disease in infancy. Pediatr Infect Dis J. 2006;25:80–1.
Tellería-Orriols JJ, García-Salido A, Varillas D, Serrano-González A, Casado-Flores J. TLR2-TLR4/CD14 polymorphisms and predisposition to severe invasive infections by Neisseria meningitidis and Streptococcus pneumoniae. Med Intensiva. 2014;38:356–62.
Smirnova I, Mann N, Dols A, Derkx HH, Hibberd ML, Levin M, Beutler B. Assay of locus-specific genetic load implicates rare toll-like receptor 4 mutations in meningococcal susceptibility. Proc Natl Acad Sci. 2003;100:6075–6080.
Read RC, Pullin J, Gregory S, Borrow R, Kaczmarski EB, Giovine FS, Dower SK, Cannings C, Ailson AG. A functional polymorphism of toll-like receptor 4 is not associated with likelihood or severity of meningococcal disease. J Infect Dis. 2001;184:640–2.
Biebl A, Muendlein A, Kazakbaeva Z, Heuberger S, Sonderegger G, Drexel H, Lau S, Nickel R, Kabesch M, Simma B. CD14 C-159T and toll-like receptor 4 Asp299Gly polymorphisms in surviving meningococcal disease patients. PLoS One. 2009;4:7374.
Allen A, Obaro S, Bojang K, Awomoyi AA, Greenwood BM, Whittle H, et al. Variation in toll-like receptor 4 and susceptibility to group A meningococcal meningitis inGambian children. Pediatr Infect Dis J. 2003;22:1018–9.
van Well GT, Sanders MS, Ouburg S, Kumar V, van Furth AM, Morre SA. Single nucleotide polymorphisms in pathogen recognition receptor genes are associated with susceptibility to meningococcal meningitis in a pediatric cohort. PLoS One. 2013;8:20.
Sanders MS, van Well GT, Ouburg S, Lundberg PS, van Furth AM, Morre SA. Single nucleotide polymorphisms in TLR9 are highly associated with susceptibility to bacterial meningitis in children. Clin Infect Dis. 2011;52:475–80.
Zhang, PZhang N, Liu L, Zheng K, Zhu L, Zhu J, Cao L, Jiang Y, Liu G, He Q. Polymorphisms of toll-like receptors 2 and 9 and severity and prognosis of bacterial meningitis in Chinese children. Sci Rep. 2017;7:42796.
Lundbo LF, Harboe ZB, Clausen LN, Hollegaard MV, Sorensen HT, Hougaard DM, Konradsen HB, Norgaard M, Benfield T. Genetic variation in NFKBIE is associated with increased risk of pneumococcal meningitis in children. BioMedicine. 2016;3:93–9.
Chapman SJ, Khor CC, Vannberg FO, Rautanen A, Segal S, Moore CE, Davies RJ, Day NP, Peshu N, Crook DW, Berkley JA, Williams TN, Scott AJ, Hill AVS. NFKBIZ polymorphisms and susceptibility to pneumococcal disease in European and African populations. Genes Immun. 2010;11:319–25.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsibile Editor: Thiago Mattar Cunha.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gowin, E., Januszkiewicz-Lewandowska, D. Genes and their single nucleotide polymorphism involved in innate immune response in central nervous system in bacterial meningitis: review of literature data. Inflamm. Res. 67, 655–661 (2018). https://doi.org/10.1007/s00011-018-1158-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-018-1158-3