
Abstract
Left ventricular hypertrophy is a significant independent risk factor for sudden cardiac death, dysrhythmia, heart failure, ventricular ischemia, and coronary heart disease. However, the pathophysiological mechanisms which lead to the development of cardiac hypertrophy and eventually heart failure are not completely understood. Although the specific steps that lead to hypertrophy are not altogether clear, it appears that oxidative stress plays a significant role. As such, the possibility of a treatment approach involving antioxidant therapies such as the naturally occurring polyphenol resveratrol is a potentially fruitful strategy that can be used to target several signaling and pathogenic events in the etiology of cardiac hypertrophy and may be useful alone or in combination with other drugs. Over the past 15 years, a great deal of evidence supporting the amelioration of hypertrophy and improvement of cardiac function by resveratrol has been documented. However, this is largely in cell-based and animal models with limited evidence from human studies. Clinical trials utilizing resveratrol as a supplementary therapy in heart failure patients are just underway, and much is left to be understood regarding the distinct cellular mechanism of action of this compound in physiological and pathological states. Thus modulation of oxidative stress pathways and cardiac function by resveratrol remains an exciting and undoubtedly rewarding area of research with important clinical implications.
Similar content being viewed by others


Keywords
1 Introduction
Increased hemodynamic load on the left ventricle initiates a cascade of events that leads to ventricular remodeling, cardiac hypertrophy, and eventually heart failure [1]. This ventricular remodeling is a myocardial response to various stresses and involves gradual and progressive changes in chamber architecture, cardiomyocyte phenotype, as well as non-myocytic components [2, 3]. Clinical conditions that produce the pressure-overload stimulus include essential hypertension, aortic stenosis, aortic coarctation, and drug-induced hypertension. The pattern of ventricular remodeling and hypertrophy is dependent upon the type of cardiac stress. In the case of pressure overload, the left ventricle responds with concentric hypertrophy in contrast to the eccentric hypertrophy which occurs in response to volume overload. The process of hypertrophy is initially considered to be an adaptive response because it serves to normalize wall stress and helps to mechanically compensate heart function. However, with progression of the stimulus, the initial compensatory hypertrophy can lead to maladaptive responses that can eventually lead to heart failure. Although the exact point at which hypertrophy becomes maladaptive is difficult to identify, there may be a clear delineation of compensated and decompensated phases of hypertrophy [4–6]. Alternatively, it has also been proposed that the initial hypertrophic process involves a combination of both adaptive and maladaptive cardiac processes and that initiation of any pathological hypertrophy will eventually compromise heart function [7].
Left ventricular hypertrophy is recognized as a major independent risk factor for cardiovascular morbidity and mortality from sudden cardiac death, dysrhythmia, heart failure, ventricular ischemia, and coronary heart disease [6, 8–11]. A growing body of evidence suggests that oxidative stress, which occurs when the generation of reactive oxygen species (ROS) exceeds the cell’s intrinsic antioxidant defenses, is an important contributor to the development and progression of cardiac hypertrophy, in part by activating multiple intracellular signaling pathways [12]. Therefore, ameliorating oxidative stress has been proposed as a potential strategy for preventing and/or treating pathological cardiac hypertrophy and heart failure [13, 14]. Our current understanding of the pathophysiology of cardiac hypertrophy as well as potential new therapies targeting oxidative stress will be reviewed herein.
2 Physiological Process of Ventricular Hypertrophy
It is generally accepted that mechanical stress begins the cascade of events that leads to ventricular remodeling. However, it is important to note that in vivo, there is rarely, if ever, an isolated increase in pressure independent of any neurohormonal activation. As a result, pressure overload involves mechanical as well as neurohormonal activation that then modulates the ventricular response to stress. Left ventricular hypertrophy begins with normal ventricle and myocyte function; however, with prolonged hypertrophy this situation eventually decompensates into a hypertrophied ventricle with corresponding myocyte dysfunction. These changes influence both structural and mechanical properties of the myocardial tissue and coronary circulation [1].
At the cellular level, concentric left ventricular hypertrophy is identified by an increase in cardiomyocyte cell size, parallel addition of new sarcomeres, and enhanced protein synthesis arising from alterations in gene transcription and translation [15]. In addition to changes to the cardiomyocyte, left ventricular hypertrophy is also characterized by proliferation of fibroblasts, increased generation of extracellular matrix proteins, and fibrosis [8, 16, 17]. This increased fibroblast growth in combination with the deposition of collagen and fibrosis leads to myocardial stiffening, which on its own is an important contributor to cardiac dysfunction [18, 19]. In the adult mammalian heart, α-myosin heavy chain (MHC) is the predominant isoform expressed and possesses a high ATPase activity and faster rate of contractility than the fetal β-MHC isoform. During hypertrophy, α-MHC expression is reduced, and β-MHC expression is increased which is characteristic of the re-induction of the fetal gene program [20]. Indeed, the transition from α- to β-MHC isoforms that occurs during pathological cardiac hypertrophy may play a role in the development of cardiac dysfunction by reducing myocardial contractility [19].
Generally there are three broad steps regarding the molecular mechanism of ventricular hypertrophy. Initially, heart remodeling is activated by biomechanical stress induced by increased pressure. The left ventricle is stretched during diastole and overloaded during systole which initiates both local myocardial and systemic neurohormonal responses. The mechanical stretch is sensed by myocyte membrane and sarcomere-coupled stretch receptors [21], whereas neurohormonal signals [i.e., endothelin-1 (ET-1), angiotensin-II (Ang-II), catecholamines] are sensed by G protein-coupled receptors that lead to activation of Gαq/11 proteins [22]. In the second stage of the hypertrophic process, these mechanical and neurohormonal stimuli activate intracellular signaling pathways which eventually transmit a signal to the myocyte nucleus [7, 21]. Finally these signaling pathways activate transcription factors to induce expression of particular genes in the cardiac myocyte which ultimately causes phenotypic changes in the myocytes as well as altered gene transcription that includes re-expression of the fetal gene profile [i.e., atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP)] and new cellular growth [21, 23]. Activation of specific pathways leads to beneficial and adaptive remodeling, while activation of other pathways eventually has maladaptive and harmful results [21, 24]. Ultimately the imbalance between maladaptive and beneficial signaling results in the development of pathological hypertrophy and the progression to heart failure. Since multiple molecular signaling pathways have been shown to be involved in the development of pathological hypertrophy, these pathways are becoming potential targets for cardiovascular treatments [25].
3 Transition from Compensated Hypertrophy to Heart Failure
As mentioned, the initial stimulus for hypertrophy may also stimulate maladaptive events in the cardiac tissue [2, 18, 26]. Several of the initial changes occurring at the level of the cardiomyocyte may be detrimental in left ventricular hypertrophy. For instance, the aforementioned re-expression of the fetal gene profile also induces a change in energy metabolism that results in enhanced glycolysis and a reduction in overall oxidative metabolism [27]. Rates of glycolysis are accelerated in the hypertrophied heart likely as an initial adaptive mechanism to increase ATP production in the presence of impaired oxidative metabolism [28–31]. As a result of this metabolic reprogramming, there is a decrease in myocardial ATP production, leading to energy deprivation [23, 32]. In addition, when glucose oxidation is not correspondingly increased along with glycolysis, there is an accumulation of protons and ROS [28, 33, 34]. Moreover, the reduction in fatty acid utilization itself may lead to lipid accumulation within cardiomyocytes, which has been shown to result in contractile dysfunction [35]. In addition to metabolic changes, apoptosis of cardiomyocytes is augmented in hypertensive heart disease, which leads to a reduction in contractile mass and affects contractility [36]. Other events which occur during hypertrophy such as collagen deposition may promote ventricular stiffness and diastolic dysfunction and heart failure [37–39]. As a result of these and a myriad of other changes that occur [1], it is apparent that myocardial hypertrophy in the setting of pressure overload is ultimately a maladaptive process that should be prevented or reversed in order to prevent the development of impaired cardiac contractile function and heart failure.
4 Oxidative Stress in Cardiac Hypertrophy and Heart Failure
Although the fundamental mechanisms underlying the development and progression of cardiac hypertrophy and heart failure are not fully understood, evidence from experimental models of heart failure and clinical studies demonstrates increased oxidative stress in the failing myocardium and strongly supports the concept that it plays a major role in the development of cardiac hypertrophy and progression to heart failure [40–43]. Oxidative stress is defined as the excessive production of ROS relative to the body’s natural antioxidant systems. ROS are a group of molecules including oxygen and its derivatives that are produced in all aerobic cells; this includes highly reactive free radicals such as superoxide anion (•O2 −) and hydroxyl radical (OH•), as well as compounds such as hydrogen peroxide (H2O2) that can be converted to reactive radical species [13]. Nitric oxide (NO) is an important molecule that regulates normal cardiac function by activating soluble guanylyl cyclase and synthesis of intracellular cGMP to inhibit activation of hypertrophic signaling. In the setting of oxidative stress, •O2 − can react with NO to form highly reactive species such as peroxynitrite, which has cytotoxic effects and further reduces NO bioavailability.
There are several enzymatic sources of ROS in mammalian cells including mitochondrial respiration, arachidonic acid pathway enzymes (lipoxygenase and cyclooxygenase), cytochrome p450s, xanthine oxidase, NADH/NADPH oxidases, nitric oxide synthase (NOS), peroxidases, and other hemoproteins [12]. The three key producers of ROS in the cardiovascular system are xanthine oxidase, NADH/NADPH oxidase, and NOS [12]. To combat ROS, the body has many specific and nonspecific antioxidant defense mechanisms that scavenge and degrade ROS to nontoxic molecules, including scavenging enzymes such as superoxide dismutase (SOD), glutathione peroxidase, and catalase as well as nonenzymatic antioxidants, such as vitamins E and C [44]. Under normal conditions, SOD converts •O2 − to H2O2, which is then further detoxified by catalase and glutathione peroxidase to H2O [13]. Excessive production of ROS causes cell dysfunction, protein and lipid peroxidation, and DNA damage—all of which lead to cell damage and death [12]. In addition, ROS appear to participate in several steps involved in the development and progression of myocardial remodeling and failure, including modification of proteins critical for proper contractile function [45], activation of hypertrophy signaling kinases or transcription factors [46] and apoptosis [47], as well as stimulation of cardiac fibroblast proliferation and activation of matrix metalloproteinases leading to extracellular matrix remodeling [48–50].
4.1 Cardiac Sources of Reactive Oxygen Species
Increased oxidative stress in the failing heart may be reflective of an increase in the production of ROS from many sources and also a deficiency in antioxidant capacity. The mechanisms responsible for increased oxidative stress in cardiac hypertrophy are unknown, and little is known about the mechanism by which stimuli such as mechanical strain, neurohormones, or cytokines lead to increased ROS. However, cardiac sources of ROS include cardiac myocytes, endothelial cells, and neutrophils. Within cardiac myocytes, ROS can be produced by mitochondria due to leakage of electrons from the respiratory chain which results in the conversion of oxygen to •O2 − [51]. Mitochondria from the failing heart produce more superoxide than normal mitochondria, suggesting that mitochondrial electron transport could be a predominant source of •O2 − production [52]. In addition, these mitochondria were associated with decreased complex enzyme activity, making mitochondria an important source of ROS in failing hearts and indicating a link between mitochondrial dysfunction and oxidative stress [52].
Regarding ROS production via oxidative enzymes, NADPH oxidase activity was found to be increased by stimuli relevant to the development of heart failure (i.e., mechanical stretch, Ang II, ET-1, tumor necrosis factor-α) [53]. This increased cardiac NADPH oxidase activity has also been observed in human heart failure [54]; increased xanthine oxidase expression and activity have been similarly noted [55]. In addition to these sources of ROS, uncoupled NOS has also been shown to produce ROS in a variety of pathological conditions [56, 57]. Under normal conditions, endothelial NOS (eNOS) also known as NOS3 catalyzes the formation of NO from l-arginine and oxygen. Uncoupling of NOS due to absence of the necessary cofactor tetrahydrobiopterin (BH4), for example, produces •O2 − which is known to contribute to the progression of atherosclerosis and hypertension [58, 59]. eNOS is also uncoupled and figures importantly in pathological remodeling and progression to heart failure in response to chronic pressure overload in mice [60].
In addition to being a potential source of ROS during cardiac hypertrophy, mitochondria themselves can also be damaged by ROS. As ROS damage mitochondrial components, the •O2 − produced within the mitochondria is unable to pass through the membranes, causing still more damage to this organelle, thus compromising mitochondrial function [61]. Increased ROS generation in failing hearts was associated with mitochondrial damage and dysfunction with increased lipid peroxidation in the mitochondria and reduced oxidative capacity [62]. This establishes a vicious cycle in which mitochondrial functional decline promotes further ROS generation, more mitochondrial damage, and cellular injury.
5 Hypertrophic Signaling Pathways
A number of complex signaling cascades have been identified that regulate the hypertrophic process in the heart. There appear to be distinct signaling cascades that distinguish physiological versus pathological hypertrophy (see [63] for review of physiological hypertrophy). At a molecular level, pathological cardiac hypertrophy is mediated by several molecular growth signals and induction of fetal gene expression programs (for proteins involved in cardiac contractility and calcium handling) [22, 63]. Some of the better characterized signaling pathways involved in the development of pathological cardiac hypertrophy will be discussed in the section below, with a focus on those pathways where ROS have been shown to be important in activation of these signaling pathways. Indeed, ROS are known to regulate the activity of a variety of signaling kinases and transcription factors to mediate the cardiac hypertrophic response, including calcineurin–NFAT, mitogen-activated protein kinases (MAPKs), and AMP-activated protein kinases (AMPK) among others [63].
5.1 Calcineurin–NFAT Signaling
Calcineurin is a ubiquitously expressed calcium (Ca2+)–calmodulin-regulated serine/threonine phosphatase that consists of a catalytic A and regulatory B subunit. In response to sustained increases in intracellular Ca2+ levels, Ca2+/calmodulin complexes bind to the B subunit, inducing a conformational change allowing calcineurin to dephosphorylate downstream effector proteins, most notably the members of the nuclear factor of activated T-cells (NFAT) family of transcription factors [64]. NFAT transcription factors are normally sequestered in the cytoplasm; however, upon calcineurin-mediated dephosphorylation of NFAT, these proteins rapidly translocate into the nucleus and activate gene expression [65, 66]. Calcineurin activation is observed in hypertrophied hearts following pressure overload induced by constriction of the abdominal aorta [67]. Moreover, partial inhibition of calcineurin by nonspecific immunosuppressive drugs cyclosporin A (CsA) and FK506 has been shown to prevent hypertrophy in some studies [67, 68].
Using a more targeted genetic approach, several transgenic mice have been generated and characterized in order to better define the role of calcineurin in the development and progression of cardiac hypertrophy. Mice expressing a constitutively active form of calcineurin in the heart are sufficient to induce cardiac hypertrophy (two- to threefold increase in heart size) and the expression cardiac fetal hypertrophic genes (i.e., BNP, β-MHC), a phenotype which then rapidly progresses to dilated cardiomyopathy, interstitial fibrosis, and heart failure [66]. Furthermore, targeted inhibition of calcineurin by cardiac overexpression of the inhibitory domains of Cain/Cabin-1 and A-kinase anchoring protein (AKAP) 79, which are endogenous calcineurin inhibitors, reduced cardiac hypertrophy in response to both isoproterenol infusion and pressure overload [69].
Calcineurin activity is negatively regulated by myocyte-enriched calcineurin-interacting protein (MCIP1), which is highly expressed in striated muscle and capable of binding and inhibiting the activity of calcineurin [70]. The cardiac hypertrophic response induced by aortic banding was significantly blunted in transgenic mice overexpressing the calcineurin inhibitory domain from MCIP1 [71]. Mice expressing either a cardiac-specific dominant-negative mutant of calcineurin [72] or a deletion of calcineurin Aβ [73] both displayed significantly less cardiac hypertrophy following aortic banding. Lastly, similar protection from pressure-overload-induced hypertrophy is observed in mice with targeted disruption of NFATc3, a downstream transcription factor effector of calcineurin-mediated hypertrophy [74]. Collectively these different animal models provide strong evidence that the calcineurin–NFAT pathway plays a fundamental role in regulating sustained hypertrophic growth of the heart. Moreover, it is thought that activation of the calcineurin–NFAT pathway is specific to pathological hypertrophy, such as that produced by hypertension or aortic constriction, and does not contribute to the development of physiological cardiac hypertrophy (i.e., exercise), which does not lead to deleterious consequences, thus creating a distinction between signaling pathways responsible for the two different types of cardiac hypertrophy [64].
Alterations in calcium homeostasis, as a result of changes in expression and/or activity of Ca2+ handling proteins, are a common phenomenon observed in the setting of cardiac hypertrophy and heart failure [75]. Since cardiac contractile function is highly dependent upon intracellular Ca2+ concentration, modifications in Ca2+ handling proteins can have a profound effect on cardiac contractility and function [76]. Oxidative stress plays a major role in the indirect regulation of intracellular Ca2+ homeostasis and in general is thought to lead to a rapid increase in cytosolic Ca2+ levels. Both the sarcoplasmic reticulum ATPase (SERCA2), an ATP-dependent Ca2+ pump that transports Ca2+ from the cytoplasm into the endoplasmic/sarcoplasmic reticulum, and the ryanodine receptor (RyR) SR Ca2+ release channels are redox sensitive [45]. ROS have been shown to reduce expression and/or activity of the SERCA2A [77] and increase the activation of the RyR [78]. Indeed, SERCA protein expression and activity are found to be decreased in pressure-overload-induced cardiac hypertrophy [79]. As well, oxidants have been shown to stimulate reverse mode of the sarcolemmal sodium–calcium exchanger (NCX) to facilitate Ca2+ entry into the cell [45]. Therefore, together these mechanisms may lead to an overall increase in intracellular Ca2+ levels that then may result in activation of the calcineurin–NFAT signaling pathway to trigger hypertrophy of the myocardium [80].
Since the cardiac myocyte is constantly beating with cyclic Ca2+ transients mediating excitation–contraction coupling, it is likely that Ca2+-dependent activation of the calcineurin–NFAT pathway is more complex than in other cell types where sustained increases in total intracellular Ca2+ are sufficient to trigger activation of this pathway [81]. More recently, it has been proposed the existence of specialized cellular microdomains where Ca2+ concentration is locally regulated and sensed by Ca2+-dependent proteins localized within these microdomains [81]. Transient receptor potential (TRP) channels, which are G protein-coupled receptor (GPCR)-operated Ca2+ channels that are located on the sarcolemma and membrane of the SR/ER, are now emerging as potential key regulators of Ca2+ entry during hypertrophy [82]. Indeed, TRP channel expression and activity are upregulated in pathological hypertrophy and heart failure [83–85]. Transgenic mice overexpressing various forms of TRP channels (i.e., TRPC3 and TRPC6) display marked ventricular hypertrophy at baseline and are more sensitive to pressure-overload-induced hypertrophy, which is associated with increased calcineurin–NFAT activity [85, 86]. Moreover, targeted disruption of the calcineurin Aβ gene blocked the exaggerated hypertrophic response in TRPC3 transgenic mice when subjected to TAC [86], supporting a link between TRPC and activation of the calcineurin–NFAT pathway. ROS generated during pressure-overload hypertrophy may also contribute to activation of TRP channels in this pathological setting [87]. Collectively these data suggest that TRP channels may play an important role in mediating pathological cardiac hypertrophy, likely by increasing local Ca2+ concentrations in microdomains resulting in activation of the calcineurin–NFAT signaling pathway and subsequent gene expression.
5.2 Mitogen-Activated Protein Kinases (MAPKs)
In general, mitogen-activated protein kinases (MAPKs) are divided into three classes based on the terminal kinase in the pathway: the extracellular signal-regulated kinases (ERKs), the c-Jun amino-terminal kinase (JNKs), and the p38 MAPKs [88–90]. There is evidence for activation of all three families of MAPKs in cultured cardiac myocytes in response to hypertrophic stimuli (i.e., mechanical stress, GPCR agonists, Ca2+) [91–93], in the experimental pressure-overloaded heart [94, 95], and in the human failing heart [96]. Although these MAPKs have been largely considered to be pro-hypertrophic, the precise role of each of these MAPKs in the pathophysiology of cardiac hypertrophy and whether they are necessary mediators and/or modulators of the hypertrophic process remains to be clearly identified. However, early during the development of cardiac hypertrophy, it has been thought that ROS may contribute to activation of each these MAPK signaling pathways [97]. As these kinases are considered pro-hypertrophic, antioxidant therapies that target these signaling pathways may also prove to be advantageous in the treatment of pathological cardiac hypertrophy and the progression to heart failure.
5.3 Extracellular Signal-Regulated Kinases (ERKs)
ERK1/2 are ubiquitously expressed protein kinases that are activated in the setting of pathological cardiac hypertrophy and heart failure [63, 90, 98, 99]. In cultured cardiac myocytes, ERK1/2 are activated in response to pro-hypertrophic agonist stimulation, oxidative stress, or mechanical loading [91, 97, 100, 101]. Furthermore, activation of ERK 1/2 is required for increased protein synthesis in isolated cardiac myocytes in response to hypertrophic stimuli, such as ET-1 and α-adrenergic agonists [102]. Consistent with these in vitro findings, expression of a dominant-negative mutant of Raf-1, an upstream kinase of Erk1/2, inhibited the activation of ERK1/2 and blunted development of pressure-overload-induced cardiac hypertrophy [103]. Interestingly, mice with expression of cardiac-specific constitutively active MEK1, an immediate upstream MAPK kinase of ERK1/2, led to the development concentric physiological hypertrophy associated with enhanced cardiac systolic function without decompensation over time [104]. Furthermore, neither a global deletion of ERK1 or ERK1 −/− and ERK2 +/− mice led to a reduction in cardiac hypertrophy in mice subjected to TAC [105], suggesting that ERK1/2 may be sufficient, but not critical for mediating pathological pressure-overload-induced cardiac hypertrophy.
5.4 c-Jun Amino-Terminal Kinase (JNKs)
The JNK family consists of at least ten isoforms derived from three mammalian genes: JNK1, JNK2, and JNK3 [106]. JNK is rapidly activated in vitro in response to multiple cellular stresses, including mechanical stress, oxidative stress, and proinflammatory cytokines [106]. Activation of JNK is mediated via phosphorylation by MAPK kinase 4 (MEK4) and MEK7, which in turn are activated by MAPK kinase kinase (MEKK1) [107, 108]. Although several in vitro studies suggest that JNKs may contribute to the regulation of pathological hypertrophy [109, 110], data from in vivo studies have proven inconclusive. Studies in transgenic mouse models with blunted JNK activation have provided conflicting results with some showing that cardiac hypertrophy in response to pressure overload is either attenuated [111] or enhanced [112]. Moreover, TAC in mice with selective deletions of JNK1, JNK2, or JNK3 show that the cardiac hypertrophy developed to a similar extent as in wild-type mice [113], suggesting that either JNK isoforms perform similar functions and are redundant or that JNK activation may not be required for cardiac growth. At present, the exact role of JNK in the development of pathological cardiac hypertrophy remains unclear.
5.5 p38 MAPKs
The p38 MAPK family is an important regulator of a diverse array of biological functions including cell growth, cell proliferation, metabolism, and cell death [108, 114]. Of the four p38 isoforms, it appears that only p38α and p38β are expressed in the heart. Similar to JNK, p38 is a stress-activated protein kinase that is activated by a multitude of external stimuli including cytokines, oxidative stress, osmotic stress, and growth factors among others (see [63] for review). In vitro studies have largely supported a key role for p38 in promoting cardiac growth and hypertrophy with small molecular inhibitors of p38 and dominant-negative p38 adenovirus inhibiting hypertrophic growth [115]. Similar to the MAPKs described above, myocardial p38 activity is found to be increased by pressure overload [95] and ET-1/phenylephrine stimulation [116, 117]. Despite these findings, targeted activation of p38 in the heart by transgenic overexpression of upstream kinases MKK3 or MKK6 did not produce a significant degree of cardiac hypertrophy in the basal state. However, these mice did have increased interstitial fibrosis and ventricular wall thinning and died prematurely at 7–9 weeks as a consequence of heart failure [118], suggesting that p38 may be important in mediating late-stage ventricular remodeling as opposed to hypertrophy. Furthermore, studies in cardiac-specific p38 dominant-negative transgenic mice (DN-p38) showed that loss of p38 activity either had no effect on the development of cardiac hypertrophy [119] or enhanced the hypertrophic response [120] to pressure overload by aortic banding. In support of the latter finding, p38 MAPK has been shown to inhibit NFAT-transcriptional activity and nuclear translocation by directly phosphorylating NFAT [120, 121], suggesting that p38 may in fact be a negative regulator of hypertrophy. As several studies performed by different groups even in the same transgenic mice have given contradictory results, the precise role of p38 in the development and progression of cardiac hypertrophy and heart failure remains unclear.
5.6 AMP-Activated Protein Kinase (AMPK)
AMPK is a key metabolic sensing serine/threonine kinase that is activated by cellular and metabolic stresses that deplete ATP (i.e., ischemia). AMPK responds to increases in the AMP/ATP ratio by activating energy-producing metabolic pathways and inhibiting energy-consuming pathways [122]. The exact role of AMPK in cardiac hypertrophy has not yet been clearly defined. Protein translation and synthesis are known to be necessary mediators of increased myocardial cell size, and pharmacological activation of AMPK (i.e., AICAR, resveratrol) has been shown to inhibit protein synthesis associated with cardiac hypertrophy via numerous molecular pathways (see [122, 123] for review). Thus, while AMPK may act as a negative regulator of cardiac hypertrophy [124–127], it is not certain if inactivation of AMPK is a necessary step of the hypertrophic process or if it simply creates a permissive environment for cardiac growth. Supporting this idea, recent evidence has shown that impaired AMPK activity makes the heart more susceptible to pro-hypertrophic stimuli, such as hemodynamic overload [128–130]. In the SHR, increased oxidative stress [specifically the lipid peroxidation by-product, 4-hydroxy-2-nonenal (HNE)] leads to a reduction in AMPK activity via inhibition of its upstream activating kinase LKB1 [128], and this was associated with the development of cardiac hypertrophy. Consistent with studies in isolated myocytes, impaired AMPK activity was associated with activation of the mTOR/p70S6 kinase pathway that regulates protein synthesis in the heart [128]. Furthermore, cardiomyocyte-specific deletion of LKB1 results in left ventricular hypertrophy [131], suggesting that modification of LKB1 activity may contribute to the hypertrophic process. Although some studies have shown that the activation of AMPK is associated with the development of pressure-overload-induced cardiac hypertrophy [132], this is likely due to the heart becoming energetically compromised in the later stages of the disease and requiring AMPK to restore depleted ATP levels. Therefore, AMPK may play dual roles in the cardiac hypertrophic process whereby early in the disease, inactivation of AMPK may be necessary for cardiac growth, whereas activation of AMPK occurs during the later stages in order to maintain adequate ATP supply to the heart (see [122] for review).
5.7 JAK–STAT Pathway
Janus kinase (JAK) proteins are a family of cytosolic tyrosine kinases associated with the intracellular domain of membrane-bound receptors, which act to transduce signals from extracellular ligands such as cytokines, growth factors, and hormones to the nucleus to elicit cellular responses [133]. The JAK family of proteins rapidly transduces signals by recruitment of signal transducers and activators of transcription pathway (STAT) transcription factors. The JAK–STAT pathway plays a critical role in cardiac hypertrophy and the transition from hypertrophy to failure as well as mediates signal transduction from gp130 cytokine receptor [134] and GPCR [135] to the nucleus. Gp130 is a promiscuous receptor for several cytokines including interleukin 6/11 and transduces its signal mainly through induction of STAT3. Specifically, STAT3 is translocated to the nucleus in response to gp130 activation, which results in the induction of genes involved in hypertrophy [136]. Overexpression of STAT3 is sufficient to induce cardiomyocyte hypertrophy under both in vitro [137] and in vivo [138] settings. Transgenic mice with a deletion of gp130 in the myocardium have reduced STAT3 activity and display normal cardiac structure and function at baseline; however, these mice fail to develop compensatory hypertrophy following acute pressure overload and develop heart failure [139]. Therefore, the JAK–STAT pathway may be a novel therapeutic target for developing agents that prevent the development and/or progression of pathological cardiac hypertrophy.
The signaling pathways involved in cardiac hypertrophy are numerous and complex. The generation and characterization of transgenic rodent models have allowed investigators to better delineate the potential molecular mechanisms responsible for mediating distinct forms of cardiac growth. However, future research is still needed to clearly identify the pathways that are most critical for the development of pathological cardiac hypertrophy, and in doing so this may provide new targets for drug discovery in the management and treatment of cardiac hypertrophy and heart failure.
6 Antioxidants: Implications for Therapy
Since oxidative stress has been strongly implicated in the pathophysiology of cardiac hypertrophy induced by pressure overload, antioxidant therapies have received much attention as a potential therapeutic strategy to prevent and/or regress cardiac hypertrophy and/or prevent the transition to heart failure. To date, this therapeutic approach using antioxidants such as vitamin E and n-acetylcysteine, while having had some success in experimental animal models, has not successfully translated to the clinic for use in humans [140]. However, the natural polyphenol resveratrol that is found in a number of dietary food sources is emerging as a potential novel new therapy that may be a treatment option for those with cardiovascular disease, in particular cardiac hypertrophy.
6.1 Vitamin E
The antioxidant vitamin E is a naturally occurring lipid-soluble vitamin found in a variety of food sources, including nuts and sunflower seeds, and is a nonenzymatic part of the cell’s intrinsic antioxidant system to counter the accumulation of ROS [141]. Indeed, symptoms in humans with a vitamin E deficiency further support that vitamin E plays an important role in protecting membranes and the nervous system from oxidative stress [142]. Of the various forms of vitamin E, α-tocopherol is the most abundant and biologically active form of vitamin E, with the acetate and synthetic forms of α-tocopherol being the primary constituents of vitamin E supplements [141, 143]. Vitamin E is a potent peroxyl radical scavenger that breaks radical-propagated chain reactions and due to its lipid solubility is present in cell membranes and plays an integral role in protecting cell membranes and plasma lipoproteins from lipid peroxidation [142, 143]. Indeed, peroxyl radicals (ROO−) react at a rate 1,000 times faster with vitamin E than with polyunsaturated fatty acids [144].
In experimental models, treatment with vitamin E has been shown to reduce oxidative stress and prevent the development of cardiac hypertrophy and heart failure [145]. In the guinea pig model of pressure overload, chronic vitamin E treatment improved cardiac function and blunted the progression of heart failure over 20 weeks. Interestingly, however, this occurred in the absence of improvements in hypertrophy [145]. In contrast, clinical studies in humans have produced conflicting results regarding the cardiovascular benefit of vitamin E supplementation with some studies showing a significant reduction in cardiovascular risk [146–151], as well as a number of studies having failed to show a benefit to vitamin E supplementation [152–155]. Therefore, simply supplementing the antioxidant defenses of the heart using vitamin E may be insufficient to prevent cardiac hypertrophy and cardiovascular disease.
6.2 N-Acetylcysteine
N-acetylcysteine is a widely used thiol-containing antioxidant that is a pharmacological precursor of l-cysteine [156]. Reduced thiols are molecules with a sulfhydryl group whose biological properties include scavenging of oxygen free radicals, acting as cofactors for enzymatic reactions and potentiating the half-life and activity of NO by forming NO adducts (S-nitrosothiols) which are more stable than NO itself [157, 158]. Oral treatment with N-acetylcysteine (500 mg/kg/day) administered in the drinking water for 7 days led to a marked reduction in left ventricular weight induced by 2 weeks of abdominal aortic constriction in mice [159], which is consistent with a major role for ROS in the generation of pressure-overload-induced left ventricular hypertrophy in vivo. Interestingly, N-acetylcysteine administration (1.5 g/kg/day) to adult spontaneously hypertensive rats with established hypertension is both unable to reduce blood pressure and cardiac hypertrophy [160, 161]. Due to a lack of large-scale clinical trials, it is not clear whether N-acetylcysteine is effective in reducing cardiac hypertrophy in humans. However, N-acetylcysteine may be beneficial following myocardial infarction to limit infarct size [162, 163]. As well, N-acetylcysteine treatment has been shown to improve endothelial function and reduce systolic blood pressure in hypertensive diabetic patients via a reduction in oxidative stress and increased NO bioavailability [164]. This suggests that N-acetylcysteine can reduce oxidative stress in vivo and may be a useful antioxidant strategy to supplement current therapies in a variety of cardiovascular conditions.
7 Resveratrol as an Antioxidant
Resveratrol (3,5,4′-trihydroxystilbene) is a polyphenol that is found in a variety of berries, grapes, peanuts, and medicinal plants. Scientific interest in resveratrol has increased over the past 15 years since it has been shown to be a calorie-restriction mimetic and increase overall health in mammals [165]. Of relevance, resveratrol also appears to prevent the development of pressure-overload-induced cardiac hypertrophy [128, 166, 167] as well as show benefit in a variety of other cardiovascular diseases [168]. Indeed, resveratrol has many properties which are considered the basis of its cardiovascular effects and protection. Some of these include antioxidant, •O2 − scavenging, ischemic preconditioning, and angiogenic actions [169]. The exact mechanism of action of resveratrol in vivo has not been unequivocally determined; however, many cell components and processes are thought to be involved such as surface receptors, signaling pathways, metabolic pathways, nuclear receptors, and gene transcription and translation [170]. The antioxidant and •O2 − scavenging properties of resveratrol are of interest as they directly relate to the potential therapeutic benefits of this compound to reduce oxidative stress in the setting of cardiac hypertrophy. Some experiments suggest that resveratrol can directly scavenge •O2 − in potassium superoxide and xanthine oxidase-based antioxidant systems [171], with added evidence of direct inhibition of xanthine oxidase. Resveratrol has also been shown to inhibit NADPH oxidase and subsequently reduce •O2 − production as a mechanism for vasorelaxation in isolated rat aorta [172]. Additional evidence supports direct scavenging of ROS by resveratrol [173, 174] as well as increased expression of antioxidant enzymes such as SOD, catalase, and glutathione peroxidase [174–176]. Thus a key protective mechanism of resveratrol appears to be an augmentation of endogenous antioxidant systems in addition to direct inhibition/scavenging of ROS to counter the characteristic increase in oxidative stress during the development of cardiac hypertrophy.
In vivo experiments suggest that resveratrol is indeed a potent antioxidant via upregulation of NOS and increased NO production or by modulation of thioredoxin and heme oxygenase systems [177]. In fact, NO may be an important component in resveratrol-induced changes in intracellular redox state because NO has a greater affinity for •O2 − radicals compared to superoxide dismutase and thus can rapidly lower •O2 − concentrations [178]. In support of this concept, platelet eNOS was activated at physiologically achievable doses of resveratrol and blunted the proinflammatory pathway linked to p38 MAPK to inhibit production of ROS [179]. The proposed mechanism for increased NO production by resveratrol is thought to involve activation of the sirtuin SIRT1 protein deacetylase [180] which in turn has been shown to deacetylate eNOS at lysine residues to stimulate NO production [181, 182]. Related to this, resveratrol also activates AMPK [165, 183], which can then directly phosphorylate eNOS to stimulate NO production [184, 185]. As NO possesses vasodilatory, anti-inflammatory, anti-hypertrophic properties [186], increasing NO production may be an important pathway by which resveratrol ameliorates pathological cardiac hypertrophy.
As mentioned, mitochondria contribute importantly to the increased oxidative stress in cardiac hypertrophy. Interestingly, resveratrol has also been shown to attenuate mitochondrial oxidative damage [187], which could be another potential mechanism by which this compound is cardioprotective. Resveratrol-induced overexpression of SIRT1 attenuates mitochondrial oxidative damage in endothelial cells [188, 189], suggesting that prevention of mitochondrial oxidative damage alone could be an indirect mechanism for reduction of ROS by resveratrol. Furthermore, resveratrol also reduces mitochondrial •O2 − production in many cell types, including human coronary arterial endothelial cells which have been attributed to direct stimulation of mitochondrial antioxidant systems [189].
8 Modulation of Cell Signaling by Resveratrol
Although a currently accepted mode of action of resveratrol is as an antioxidant, this polyphenol also triggers other mechanistic pathways, which include modulation of cell signaling, apoptosis, and gene expression [190]. For example, resveratrol modulates phorbol-ester-induced signal transduction pathways, which leads to elevated COX2 expression [191] as well as other signals such as NFkB, MAP kinases, transcription factor activating protein-1 (AP-1), and ERK [192]. In addition, resveratrol interferes with many intracellular signaling pathways that regulate cell survival and apoptosis [193].
With specific relevance to cardiac pathophysiology, it is known that resveratrol reduces phenylephrine-induced hypertrophy of rat cardiac myocytes through the activation of AMPK in an AMPK kinase, LKB1-dependent manner [194]. Resveratrol acts as a negative regulator of cardiac hypertrophy via inhibition of the mTOR–p70S6 kinase protein synthesis pathway [195, 196]. Furthermore, resveratrol has been shown to inhibit calcineurin activity, NFAT translocation to the nucleus, and NFAT-dependent transcription in phenylephrine-induced hypertrophy of isolated rat cardiac myocytes [194]. As the calcineurin–NFAT signaling pathway is a major contributor to the development of pathological hypertrophy, resveratrol’s ability to suppress activation of this pathway may be an important mechanism by which resveratrol mediates its anti-hypertrophic effects.
As discussed, it is widely accepted that oxidative stress due to excessive production of ROS is a prominent factor in triggering the events that result in cardiomyocyte death. Cardiac SIRT1 is the mammalian ortholog of the silent information regulator 2 (Sir 2) family and is upregulated in response to oxidative stress [197]. Resveratrol has also been shown to be an important activator of SIRT1 [198], and in neonatal rat ventricular cardiomyocytes with simulated ischemia–reperfusion injury, resveratrol-induced SIRT1 activation and overexpression protected cardiomyocytes from oxidative injury, mitochondrial dysfunction, and cell death [198]. Resveratrol-induced SIRT1 overexpression in turn affects the MAPK pathway by reducing p38 and JNK phosphorylation [198]. In a porcine coronary artery preparation, resveratrol treatment countered ET-1 enhancement of MAPK activity by directly inhibiting MAPK activity and similarly reducing JNK-1 and p38 phosphorylation, as well as phosphorylation of ERK 1/2 was also reduced [199]. In support of this finding, resveratrol pretreatment protected against oxidative stress (H2O2)-induced cell proliferation and ERK 1/2 activation in human coronary smooth muscle cells [200]. In addition, resveratrol strongly inhibited Ang-II-induced hypertrophy and produced dose-dependent reductions in Akt 1 protein kinase and ERK 1/2 phosphorylation (two mediators thought to be essentially involved in Ang-II-mediated hypertrophy) [201].
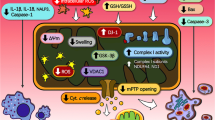
Although activation of the p38 MAPK pathway is thought to contribute to the development of cardiac hypertrophy, recent but limited studies support a beneficial role for resveratrol-mediated activation of this pathway [202]. In H9c2 embryonic rat heart-derived cells treated with H2O2 to induce oxidative stress, resveratrol protected cells from oxidative damage, increased autophagy, and reduced peroxide-induced apoptosis [202]. All of these protective effects of resveratrol were prevented by prior treatment with a p38 MAPK inhibitor [202]. Overall resveratrol protected these cells from oxidative stress by upregulating autophagy via the p38 MAPK pathway [202]. Altogether these studies emphasize that the cellular actions of resveratrol are varied and dependent on the particular tissue or system under study (Fig. 14.1).

Schematic representation of multiple cell signaling events modulated by resveratrol during the development of cardiac hypertrophy. Resveratrol affects several signaling events, many of which lead to a reduction in ROS and ultimately ameliorate cardiac hypertrophy. NFAT nuclear factor of activated T-cells, mTOR mammalian target of rapamycin, AMPK AMP-activated protein kinase, eNOS endothelial nitric oxide synthase, NOS nitric oxide synthase, NO nitric oxide, ROS reactive oxygen species, SIRT1 sirtuin (silent mating type information regulation 2 homolog) 1, p38: p38 mitogen-activated kinase, JNK: c-Jun N-terminal kinases, ERK extracellular signal-regulated kinase
9 Therapeutic Applications for Resveratrol in Cardiac Hypertrophy
To complement direct antioxidant effects which could attenuate oxidative stress and the progression of hypertrophy, resveratrol also modulates other aspects of the pathophysiology of cardiac hypertrophy. As increased arterial pressure is often an initiating factor for cardiac remodeling, the ability of resveratrol to prevent increases in blood pressure in vivo is protective against left ventricular hypertrophy [203]. In several rat models of hypertension, resveratrol prevented left ventricular hypertrophy due to reduced systolic blood pressure [204–206]. Resveratrol also appears to have direct effects on cardiac myocyte growth since it can prevent cardiac hypertrophy in the absence of changes in blood pressure in spontaneously hypertensive rats or rats subjected to abdominal aortic banding as a model of pressure overload [128, 166, 167, 207]. This direct effect of resveratrol on cardiomyocytes is also supported by the modulation of signaling pathways that regulate growth and protein synthesis [194, 195]. Resveratrol also protects against other hypertrophy-induced events such as preventing the reduction in eNOS and iNOS [207] and presumably preventing a subsequent reduction in NO production. In addition, resveratrol inhibits critical steps in cardiac collagen deposition such as cardiac fibroblast proliferation which is thought to involve the cardiac NO–cGMP signaling pathway [196, 208]. Overall, evidence demonstrates that resveratrol protects cardiomyocytes from oxidative stress and death via ROS reduction, increased expression of antioxidant enzymes, and mitochondrial protection [209, 210], with ultimate reduction of fibrosis and preservation of cardiac function and survival in rodent models of heart failure [211].
10 Clinical Uses of Resveratrol
Although experimental models suggest that resveratrol may be an effective therapy for cardiac hypertrophy, to date there is a paucity of published human trials in this area. One study in healthy humans showed that a supplement which included 100 mg of resveratrol reduced the oxidative and inflammatory responses normally induced by a high-fat, high-carbohydrate meal [212]. A recent study focused on patients after myocardial infarction who were given a 10 mg/day resveratrol supplement for 3 months. In this study, resveratrol appeared to improve left ventricular diastolic function and endothelial function as measured by flow-mediated dilation of the brachial artery with no change in blood pressure [213]. However, despite the limited published evidence in humans, there are several clinical trials underway with this compound, and resveratrol may be a promising new therapeutic strategy for the treatment of cardiac hypertrophy and heart failure.
11 Conclusion
A thorough grasp of the pathophysiological mechanisms underlying the development of cardiac hypertrophy and the progression to heart failure is essential considering that left ventricular hypertrophy is a significant independent risk factor for sudden cardiac death, dysrhythmia, heart failure, ventricular ischemia, and coronary heart disease [214]. Although the specific steps that lead to hypertrophy are not completely clear, it appears that oxidative stress does play an important role in this condition. It is also promising that a treatment approach involving antioxidant therapies, including the natural polyphenol resveratrol, appears to target several signaling and pathogenic events in the etiology of this condition and may well prove to be effective in the prevention and treatment of cardiac hypertrophy alone or in combination with other drugs. Although we have a great deal of evidence supporting resveratrol treatment of hypertrophy, this is largely in cell-based and animal models with limited evidence from human studies—particularly focused on oxidative stress and cardiac function. Therefore, this is a critical and undoubtedly fruitful avenue of research in the future that may have significant clinical implications.
References
Frohlich ED, Susic D (2012) Pressure overload. Heart Fail Clin 8:21–32
Opie LH, Commerford PJ, Gersh BJ et al (2006) Controversies in ventricular remodelling. Lancet 367:356–367
Pfeffer JM, Pfeffer MA, Braunwald E (1985) Influence of chronic captopril therapy on the infarcted left ventricle of the rat. Circ Res 57:84–95
Meerson FZ (1962) Compensatory hyperfunction of the heart and cardiac insufficiency. Circ Res 10:250–258
Grossman W, Jones D, McLaurin LP (1975) Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest 56:56–64
Frohlich ED, Apstein C, Chobanian AV et al (1992) The heart in hypertension. N Engl J Med 327:998–1008
Barry SP, Townsend PA (2010) What causes a broken heart–molecular insights into heart failure. Int Rev Cell Mol Biol 284:113–179
Diez J, Frohlich ED (2010) A translational approach to hypertensive heart disease. Hypertension 55:1–8
Frohlich ED (2001) Fibrosis and ischemia: the real risks in hypertensive heart disease. Am J Hypertens 14:194S–199S
Kannel WB, Castelli WP, McNamara PM et al (1972) Role of blood pressure in the development of congestive heart failure. The Framingham study. N Engl J Med 287:781–787
Dunn FG, Pringle SD (1987) Left ventricular hypertrophy and myocardial ischemia in systemic hypertension. Am J Cardiol 60:19I–22I
Cai H, Harrison DG (2000) Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87:840–844
Takimoto E, Kass DA (2007) Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 49:241–248
Tsutsui H, Kinugawa S, Matsushima S (2011) Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 301:H2181–H2190
Anversa P, Ricci R, Olivetti G (1986) Quantitative structural analysis of the myocardium during physiologic growth and induced cardiac hypertrophy: a review. J Am Coll Cardiol 7:1140–1149
Jalil JE, Doering CW, Janicki JS et al (1989) Fibrillar collagen and myocardial stiffness in the intact hypertrophied rat left ventricle. Circ Res 64:1041–1050
Camelliti P, Borg TK, Kohl P (2005) Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res 65:40–51
Weber KT, Janicki JS, Shroff SG et al (1988) Collagen remodeling of the pressure-overloaded, hypertrophied nonhuman primate myocardium. Circ Res 62:757–765
Weber KT, Brilla CG (1991) Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 83:1849–1865
Morkin E (2000) Control of cardiac myosin heavy chain gene expression. Microsc Res Tech 50:522–531
Xu J, Carretero OA, Liao TD et al (2010) Local angiotensin II aggravates cardiac remodeling in hypertension. Am J Physiol Heart Circ Physiol 299:H1328–H1338
Frey N, Olson EN (2003) Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 65:45–79
Fouad FM, Slominski JM, Tarazi RC (1984) Left ventricular diastolic function in hypertension: relation to left ventricular mass and systolic function. J Am Coll Cardiol 3:1500–1506
Hannenhalli S, Putt ME, Gilmore JM et al (2006) Transcriptional genomics associates FOX transcription factors with human heart failure. Circulation 114:1269–1276
Luedde M, Katus HA, Frey N (2006) Novel molecular targets in the treatment of cardiac hypertrophy. Recent Pat Cardiovasc Drug Discov 1:1–20
Diez J (2009) Towards a new paradigm about hypertensive heart disease. Med Clin North Am 93:637–645
Ingwall JS (2009) Energy metabolism in heart failure and remodelling. Cardiovasc Res 81:412–419
Allard MF, Schonekess BO, Henning SL et al (1994) Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol 267:H742–H750
Takeyama D, Kagaya Y, Yamane Y et al (1995) Effects of chronic right ventricular pressure overload on myocardial glucose and free fatty acid metabolism in the conscious rat. Cardiovasc Res 29:763–767
Massie BM, Schaefer S, Garcia J et al (1995) Myocardial high-energy phosphate and substrate metabolism in swine with moderate left ventricular hypertrophy. Circulation 91:1814–1823
Leong HS, Brownsey RW, Kulpa JE et al (2003) Glycolysis and pyruvate oxidation in cardiac hypertrophy–why so unbalanced? Comp Biochem Physiol A Mol Integr Physiol 135:499–513
Beer M, Seyfarth T, Sandstede J et al (2002) Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol 40:1267–1274
Sambandam N, Lopaschuk GD, Brownsey RW et al (2002) Energy metabolism in the hypertrophied heart. Heart Fail Rev 7:161–173
Doenst T, Pytel G, Schrepper A et al (2010) Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res 86:461–470
Wende AR, Abel ED (2010) Lipotoxicity in the heart. Biochim Biophys Acta 1801:311–319
Gonzalez A, Lopez B, Ravassa S et al (2002) Stimulation of cardiac apoptosis in essential hypertension: potential role of angiotensin II. Hypertension 39:75–80
Berk BC, Fujiwara K, Lehoux S (2007) ECM remodeling in hypertensive heart disease. J Clin Invest 117:568–575
Houser SR, Piacentino V 3rd, Weisser J (2000) Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol 32:1595–1607
Weber KT (1989) Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 13:1637–1652
Belch JJ, Bridges AB, Scott N et al (1991) Oxygen free radicals and congestive heart failure. Br Heart J 65:245–248
Hill MF, Singal PK (1996) Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol 148:291–300
Hill MF, Singal PK (1997) Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation 96:2414–2420
Mallat Z, Philip I, Lebret M et al (1998) Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 97:1536–1539
Chen AF, Chen DD, Daiber A et al (2012) Free radical biology of the cardiovascular system. Clin Sci (Lond) 123:73–91
Zima AV, Blatter LA (2006) Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71:310–321
Sabri A, Hughie HH, Lucchesi PA (2003) Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid Redox Signal 5:731–740
Cesselli D, Jakoniuk I, Barlucchi L et al (2001) Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res 89:279–286
Rajagopalan S, Meng XP, Ramasamy S et al (1996) Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest 98:2572–2579
Kinugawa S, Tsutsui H, Hayashidani S et al (2000) Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res 87:392–398
Spinale FG, Coker ML, Thomas CV et al (1998) Time-dependent changes in matrix metalloproteinase activity and expression during the progression of congestive heart failure: relation to ventricular and myocyte function. Circ Res 82:482–495
Montezano AC, Touyz RM (2012) Molecular mechanisms of hypertension-reactive oxygen species and antioxidants: a basic science update for the clinician. Can J Cardiol 28:288–295
Sawyer DB, Colucci WS (2000) Mitochondrial oxidative stress in heart failure: “oxygen wastage” revisited. Circ Res 86:119–120
Landmesser U, Dikalov S, Price SR et al (2003) Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111:1201–1209
Heymes C, Bendall JK, Ratajczak P et al (2003) Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 41:2164–2171
Cappola TP, Kass DA, Nelson GS et al (2001) Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation 104:2407–2411
Sullivan JC, Pollock JS (2006) Coupled and uncoupled NOS: separate but equal? Uncoupled NOS in endothelial cells is a critical pathway for intracellular signaling. Circ Res 98:717–719
Kwon SH, Pimentel DR, Remondino A et al (2003) H(2)O(2) regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol 35:615–621
Cosentino F, Patton S, d’Uscio LV et al (1998) Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensive rats. J Clin Invest 101:1530–1537
Ozaki M, Kawashima S, Yamashita T et al (2002) Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J Clin Invest 110:331–340
Takimoto E, Champion HC, Li M et al (2005) Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest 115:1221–1231
Ballinger SW, Patterson C, Yan CN et al (2000) Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ Res 86:960–966
Ide T, Tsutsui H, Hayashidani S et al (2001) Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 88:529–535
Bernardo BC, Weeks KL, Pretorius L et al (2010) Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther 128:191–227
Wilkins BJ, Dai YS, Bueno OF et al (2004) Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res 94:110–118
Aramburu J, Rao A, Klee CB (2000) Calcineurin: from structure to function. Curr Top Cell Regul 36:237–295
Molkentin JD, Lu JR, Antos CL et al (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93:215–228
Shimoyama M, Hayashi D, Takimoto E et al (1999) Calcineurin plays a critical role in pressure overload-induced cardiac hypertrophy. Circulation 100:2449–2454
Sussman MA, Lim HW, Gude N et al (1998) Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science 281:1690–1693
De Windt LJ, Lim HW, Bueno OF et al (2001) Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc Natl Acad Sci USA 98:3322–3327
Kingsbury TJ, Cunningham KW (2000) A conserved family of calcineurin regulators. Genes Dev 14:1595–1604
Hill JA, Rothermel B, Yoo KD et al (2002) Targeted inhibition of calcineurin in pressure-overload cardiac hypertrophy. Preservation of systolic function. J Biol Chem 277:10251–10255
Zou Y, Hiroi Y, Uozumi H et al (2001) Calcineurin plays a critical role in the development of pressure overload-induced cardiac hypertrophy. Circulation 104:97–101
Bueno OF, Wilkins BJ, Tymitz KM et al (2002) Impaired cardiac hypertrophic response in Calcineurin Abeta -deficient mice. Proc Natl Acad Sci USA 99:4586–4591
Wilkins BJ, De Windt LJ, Bueno OF et al (2002) Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol 22:7603–7613
Bito V, Heinzel FR, Biesmans L et al (2008) Crosstalk between L-type Ca2+ channels and the sarcoplasmic reticulum: alterations during cardiac remodelling. Cardiovasc Res 77:315–324
Ermak G, Davies KJ (2002) Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol 38:713–721
Castilho RF, Carvalho-Alves PC, Vercesi AE et al (1996) Oxidative damage to sarcoplasmic reticulum Ca(2+)-pump induced by Fe2+/H2O2/ascorbate is not mediated by lipid peroxidation or thiol oxidation and leads to protein fragmentation. Mol Cell Biochem 159:105–114
Xu L, Eu JP, Meissner G et al (1998) Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279:234–237
de la Bastie D, Levitsky D, Rappaport L et al (1990) Function of the sarcoplasmic reticulum and expression of its Ca2(+)-ATPase gene in pressure overload-induced cardiac hypertrophy in the rat. Circ Res 66:554–564
Wilkins BJ, Molkentin JD (2004) Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun 322:1178–1191
Houser SR, Molkentin JD (2008) Does contractile Ca2+ control calcineurin-NFAT signaling and pathological hypertrophy in cardiac myocytes? Sci Signal 1:31
Eder P, Molkentin JD (2011) TRPC channels as effectors of cardiac hypertrophy. Circ Res 108:265–272
Seth M, Zhang ZS, Mao L et al (2009) TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res 105:1023–1030
Bush EW, Hood DB, Papst PJ et al (2006) Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem 281:33487–33496
Kuwahara K, Wang Y, McAnally J et al (2006) TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest 116:3114–3126
Nakayama H, Wilkin BJ, Bodi I et al (2006) Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J 20:1660–1670
Poteser M, Graziani A, Rosker C et al (2006) TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem 281:13588–13595
Clerk A, Sugden PH (1999) Activation of protein kinase cascades in the heart by hypertrophic G protein-coupled receptor agonists. Am J Cardiol 83:64H–69H
Pearson G, Robinson F, Beers Gibson T et al (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22:153–183
Muslin AJ (2008) MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin Sci (Lond) 115:203–218
Yamazaki T, Tobe K, Hoh E et al (1993) Mechanical loading activates mitogen-activated protein kinase and S6 peptide kinase in cultured rat cardiac myocytes. J Biol Chem 268:12069–12076
Sadoshima J, Qiu Z, Morgan JP et al (1995) Angiotensin II and other hypertrophic stimuli mediated by G protein-coupled receptors activate tyrosine kinase, mitogen-activated protein kinase, and 90-kD S6 kinase in cardiac myocytes. The critical role of Ca(2+)-dependent signaling. Circ Res 76:1–15
Komuro I, Kudo S, Yamazaki T et al (1996) Mechanical stretch activates the stress-activated protein kinases in cardiac myocytes. FASEB J 10:631–636
Esposito G, Prasad SV, Rapacciuolo A et al (2001) Cardiac overexpression of a G(q) inhibitor blocks induction of extracellular signal-regulated kinase and c-Jun NH(2)-terminal kinase activity in in vivo pressure overload. Circulation 103:1453–1458
Takeishi Y, Huang Q, Abe J et al (2001) Src and multiple MAP kinase activation in cardiac hypertrophy and congestive heart failure under chronic pressure-overload: comparison with acute mechanical stretch. J Mol Cell Cardiol 33:1637–1648
Cook SA, Sugden PH, Clerk A (1999) Activation of c-Jun N-terminal kinases and p38-mitogen-activated protein kinases in human heart failure secondary to ischaemic heart disease. J Mol Cell Cardiol 31:1429–1434
Tanaka K, Honda M, Takabatake T (2001) Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J Am Coll Cardiol 37:676–685
Meng R, Pei Z, Zhang A et al (2011) AMPK activation enhances PPARalpha activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling pathway. Arch Biochem Biophys 511:1–7
Ramos JW (2008) The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol 40:2707–2719
Bogoyevitch MA, Glennon PE, Andersson MB et al (1994) Endothelin-1 and fibroblast growth factors stimulate the mitogen-activated protein kinase signaling cascade in cardiac myocytes. The potential role of the cascade in the integration of two signaling pathways leading to myocyte hypertrophy. J Biol Chem 269:1110–1119
Post GR, Goldstein D, Thuerauf DJ et al (1996) Dissociation of p44 and p42 mitogen-activated protein kinase activation from receptor-induced hypertrophy in neonatal rat ventricular myocytes. J Biol Chem 271:8452–8457
Wang L, Proud CG (2002) Ras/Erk signaling is essential for activation of protein synthesis by Gq protein-coupled receptor agonists in adult cardiomyocytes. Circ Res 91:821–829
Harris IS, Zhang S, Treskov I et al (2004) Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation 110:718–723
Bueno OF, De Windt LJ, Tymitz KM et al (2000) The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J 19:6341–6350
Purcell NH, Wilkins BJ, York A et al (2007) Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc Natl Acad Sci USA 104:14074–14079
Waetzig V, Herdegen T (2005) Context-specific inhibition of JNKs: overcoming the dilemma of protection and damage. Trends Pharmacol Sci 26:455–461
Haeusgen W, Herdegen T, Waetzig V (2011) The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur J Cell Biol 90:536–544
Rose BA, Force T, Wang Y (2010) Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev 90:1507–1546
Wang Y, Su B, Sah VP et al (1998) Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. J Biol Chem 273:5423–5426
Choukroun G, Hajjar R, Kyriakis JM et al (1998) Role of the stress-activated protein kinases in endothelin-induced cardiomyocyte hypertrophy. J Clin Invest 102:1311–1320
Choukroun G, Hajjar R, Fry S et al (1999) Regulation of cardiac hypertrophy in vivo by the stress-activated protein kinases/c-Jun NH(2)-terminal kinases. J Clin Invest 104:391–398
Liang Q, Bueno OF, Wilkins BJ et al (2003) c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J 22:5079–5089
Tachibana H, Perrino C, Takaoka H et al (2006) JNK1 is required to preserve cardiac function in the early response to pressure overload. Biochem Biophys Res Commun 343:1060–1066
Zarubin T, Han J (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res 15:11–18
Liang Q, Molkentin JD (2003) Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol 35:1385–1394
Ueyama T, Kawashima S, Sakoda T et al (1999) Endothelin-1 activates p38 mitogen-activated protein kinase via endothelin-A receptor in rat myocardial cells. Mol Cell Biochem 199:119–124
Clerk A, Michael A, Sugden PH (1998) Stimulation of the p38 mitogen-activated protein kinase pathway in neonatal rat ventricular myocytes by the G protein-coupled receptor agonists, endothelin-1 and phenylephrine: a role in cardiac myocyte hypertrophy? J Cell Biol 142:523–535
Liao P, Georgakopoulos D, Kovacs A et al (2001) The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci USA 98:12283–12288
Zhang S, Weinheimer C, Courtois M et al (2003) The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J Clin Invest 111:833–841
Braz JC, Bueno OF, Liang Q et al (2003) Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest 111:1475–1486
Molkentin JD (2004) Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res 63:467–475
Dyck JR, Lopaschuk GD (2006) AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol 574:95–112
Chan AY, Dyck JR (2005) Activation of AMP-activated protein kinase (AMPK) inhibits protein synthesis: a potential strategy to prevent the development of cardiac hypertrophy. Can J Physiol Pharmacol 83:24–28
Chan AY, Soltys CL, Young ME et al (2004) Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem 279:32771–32779
Horman S, Beauloye C, Vertommen D et al (2003) Myocardial ischemia and increased heart work modulate the phosphorylation state of eukaryotic elongation factor-2. J Biol Chem 278:41970–41976
Horman S, Browne G, Krause U et al (2002) Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol 12:1419–1423
Cheng SW, Fryer LG, Carling D et al (2004) Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J Biol Chem 279:15719–15722
Dolinsky VW, Chan AY, Robillard Frayne I et al (2009) Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation 119:1643–1652
Zarrinpashneh E, Beauloye C, Ginion A et al (2008) AMPKalpha2 counteracts the development of cardiac hypertrophy induced by isoproterenol. Biochem Biophys Res Commun 376:677–681
Zhang P, Hu X, Xu X et al (2008) AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension 52:918–924
Ikeda Y, Sato K, Pimentel DR et al (2009) Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J Biol Chem 284:35839–35849
Tian R, Musi N, D’Agostino J et al (2001) Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation 104:1664–1669
O’Shea JJ, Gadina M, Schreiber RD (2002) Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell 109(Suppl):S121–S131
Kunisada K, Hirota H, Fujio Y et al (1996) Activation of JAK-STAT and MAP kinases by leukemia inhibitory factor through gp130 in cardiac myocytes. Circulation 94:2626–2632
Pelletier S, Duhamel F, Coulombe P et al (2003) Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors. Mol Cell Biol 23:1316–1333
Yamauchi-Takihara K, Kishimoto T (2000) A novel role for STAT3 in cardiac remodeling. Trends Cardiovasc Med 10:298–303
Kunisada K, Tone E, Fujio Y et al (1998) Activation of gp130 transduces hypertrophic signals via STAT3 in cardiac myocytes. Circulation 98:346–352
Kunisada K, Negoro S, Tone E et al (2000) Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci USA 97:315–319
Hirota H, Chen J, Betz UA et al (1999) Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 97:189–198
Tinkel J, Hassanain H, Khouri SJ (2012) Cardiovascular antioxidant therapy: a review of supplements, pharmacotherapies, and mechanisms. Cardiol Rev 20:77–83
Saremi A, Arora R (2010) Vitamin E and cardiovascular disease. Am J Ther 17:e56–e65
Traber MG, Stevens JF (2011) Vitamins C and E: beneficial effects from a mechanistic perspective. Free Radic Biol Med 51:1000–1013
Dutta A, Dutta SK (2003) Vitamin E and its role in the prevention of atherosclerosis and carcinogenesis: a review. J Am Coll Nutr 22:258–268
Buettner GR (1993) The pecking order of free radicals and antioxidants: lipid peroxidation, alpha-tocopherol, and ascorbate. Arch Biochem Biophys 300:535–543
Dhalla AK, Hill MF, Singal PK (1996) Role of oxidative stress in transition of hypertrophy to heart failure. J Am Coll Cardiol 28:506–514
Stephens NG, Parsons A, Schofield PM et al (1996) Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 347:781–786
Boaz M, Smetana S, Weinstein T et al (2000) Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): randomised placebo-controlled trial. Lancet 356:1213–1218
Salonen JT, Nyyssonen K, Salonen R et al (2000) Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) study: a randomized trial of the effect of vitamins E and C on 3-year progression of carotid atherosclerosis. J Intern Med 248:377–386
Stampfer MJ, Hennekens CH, Manson JE et al (1993) Vitamin E consumption and the risk of coronary disease in women. N Engl J Med 328:1444–1449
Rimm EB, Stampfer MJ, Ascherio A et al (1993) Vitamin E consumption and the risk of coronary heart disease in men. N Engl J Med 328:1450–1456
Salonen RM, Nyyssonen K, Kaikkonen J et al (2003) Six-year effect of combined vitamin C and E supplementation on atherosclerotic progression: the Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) Study. Circulation 107:947–953
Yusuf S, Dagenais G, Pogue J et al (2000) Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 342:154–160
Hodis HN, Mack WJ, LaBree L et al (2002) Alpha-tocopherol supplementation in healthy individuals reduces low-density lipoprotein oxidation but not atherosclerosis: the Vitamin E Atherosclerosis Prevention Study (VEAPS). Circulation 106:1453–1459
Heart Protection Study Collaborative Group (2002) MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 360:23–33
Steinhubl SR (2008) Why have antioxidants failed in clinical trials? Am J Cardiol 101:14D–19D
Aruoma OI, Halliwell B, Hoey BM et al (1989) The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med 6:593–597
Andrews NP, Prasad A, Quyyumi AA (2001) N-acetylcysteine improves coronary and peripheral vascular function. J Am Coll Cardiol 37:117–123
Stamler JS, Simon DI, Osborne JA et al (1992) S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA 89:444–448
Byrne JA, Grieve DJ, Bendall JK et al (2003) Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res 93:802–805
Pechanova O, Zicha J, Paulis L et al (2007) The effect of N-acetylcysteine and melatonin in adult spontaneously hypertensive rats with established hypertension. Eur J Pharmacol 561:129–136
Pechanova O, Zicha J, Kojsova S et al (2006) Effect of chronic N-acetylcysteine treatment on the development of spontaneous hypertension. Clin Sci (Lond) 110:235–242
Sochman J (2002) N-acetylcysteine in acute cardiology: 10 years later: what do we know and what would we like to know?! J Am Coll Cardiol 39:1422–1428
Sochman J, Vrbska J, Musilova B et al (1996) Infarct Size Limitation: acute N-acetylcysteine defense (ISLAND trial): preliminary analysis and report after the first 30 patients. Clin Cardiol 19:94–100
Martina V, Masha A, Gigliardi VR et al (2008) Long-term N-acetylcysteine and L-arginine administration reduces endothelial activation and systolic blood pressure in hypertensive patients with type 2 diabetes. Diabetes Care 31:940–944
Baur JA, Pearson KJ, Price NL et al (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444:337–342
Thandapilly SJ, Wojciechowski P, Behbahani J et al (2010) Resveratrol prevents the development of pathological cardiac hypertrophy and contractile dysfunction in the SHR without lowering blood pressure. Am J Hypertens 23:192–196
Wojciechowski P, Juric D, Louis XL et al (2010) Resveratrol arrests and regresses the development of pressure overload- but not volume overload-induced cardiac hypertrophy in rats. J Nutr 140:962–968
Dolinsky VW, Dyck JR (2011) Calorie restriction and resveratrol in cardiovascular health and disease. Biochim Biophys Acta 1812:1477–1489
Vidavalur R, Otani H, Singal PK et al (2006) Significance of wine and resveratrol in cardiovascular disease: French paradox revisited. Exp Clin Cardiol 11:217–225
Pervaiz S, Holme AL (2009) Resveratrol: its biologic targets and functional activity. Antioxid Redox Signal 11:2851–2897
Jia Z, Zhu H, Misra BR et al (2008) EPR studies on the superoxide-scavenging capacity of the nutraceutical resveratrol. Mol Cell Biochem 313:187–194
Orallo F, Alvarez E, Camina M et al (2002) The possible implication of trans-Resveratrol in the cardioprotective effects of long-term moderate wine consumption. Mol Pharmacol 61:294–302
Soylemez S, Sepici A, Akar F (2009) Resveratrol supplementation gender independently improves endothelial reactivity and suppresses superoxide production in healthy rats. Cardiovasc Drugs Ther 23:449–458
Ungvari Z, Orosz Z, Rivera A et al (2007) Resveratrol increases vascular oxidative stress resistance. Am J Physiol Heart Circ Physiol 292:H2417–H2424
Spanier G, Xu H, Xia N et al (2009) Resveratrol reduces endothelial oxidative stress by modulating the gene expression of superoxide dismutase 1 (SOD1), glutathione peroxidase 1 (GPx1) and NADPH oxidase subunit (Nox4). J Physiol Pharmacol 60(Suppl 4):111–116
Brito PM, Mariano A, Almeida LM et al (2006) Resveratrol affords protection against peroxynitrite-mediated endothelial cell death: A role for intracellular glutathione. Chem Biol Interact 164:157–166
Hung LM, Su MJ, Chen JK (2004) Resveratrol protects myocardial ischemia-reperfusion injury through both NO-dependent and NO-independent mechanisms. Free Radic Biol Med 36:774–781
Inoue M, Sato EF, Park AM et al (2000) Cross-talk between NO and oxyradicals, a supersystem that regulates energy metabolism and survival of animals. Free Radic Res 33:757–770
Gresele P, Pignatelli P, Guglielmini G et al (2008) Resveratrol, at concentrations attainable with moderate wine consumption, stimulates human platelet nitric oxide production. J Nutr 138:1602–1608
Lagouge M, Argmann C, Gerhart-Hines Z et al (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127:1109–1122
Mattagajasingh I, Kim CS, Naqvi A et al (2007) SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA 104:14855–14860
Arunachalam G, Yao H, Sundar IK et al (2010) SIRT1 regulates oxidant- and cigarette smoke-induced eNOS acetylation in endothelial cells: role of resveratrol. Biochem Biophys Res Commun 393:66–72
Um JH, Park SJ, Kang H et al (2010) AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 59:554–563
Chen ZP, Mitchelhill KI, Michell BJ et al (1999) AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett 443:285–289
Robich MP, Osipov RM, Nezafat R et al (2010) Resveratrol improves myocardial perfusion in a swine model of hypercholesterolemia and chronic myocardial ischemia. Circulation 122:S142–S149
Chatterjee A, Black SM, Catravas JD (2008) Endothelial nitric oxide (NO) and its pathophysiologic regulation. Vascul Pharmacol 49:134–140
Page MM, Robb EL, Salway KD et al (2010) Mitochondrial redox metabolism: aging, longevity and dietary effects. Mech Ageing Dev 131:242–252
Kao CL, Chen LK, Chang YL et al (2010) Resveratrol protects human endothelium from H(2)O(2)-induced oxidative stress and senescence via SirT1 activation. J Atheroscler Thromb 17:970–979
Ungvari Z, Labinskyy N, Mukhopadhyay P et al (2009) Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol 297:H1876–H1881
Kovacic P, Somanathan R (2010) Multifaceted approach to resveratrol bioactivity: focus on antioxidant action, cell signaling and safety. Oxid Med Cell Longev 3:86–100
Lin HY, Tang HY, Davis FB et al (2011) Resveratrol and apoptosis. Ann N Y Acad Sci 1215:79–88
Kundu JK, Shin YK, Surh YJ (2006) Resveratrol modulates phorbol ester-induced pro-inflammatory signal transduction pathways in mouse skin in vivo: NF-kappaB and AP-1 as prime targets. Biochem Pharmacol 72:1506–1515
Fulda S, Debatin KM (2006) Resveratrol modulation of signal transduction in apoptosis and cell survival: a mini-review. Cancer Detect Prev 30:217–223
Chan AY, Dolinsky VW, Soltys CL et al (2008) Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J Biol Chem 283:24194–24201
Hannan RD, Jenkins A, Jenkins AK et al (2003) Cardiac hypertrophy: a matter of translation. Clin Exp Pharmacol Physiol 30:517–527
Olson ER, Naugle JE, Zhang X et al (2005) Inhibition of cardiac fibroblast proliferation and myofibroblast differentiation by resveratrol. Am J Physiol Heart Circ Physiol 288:H1131–H1138
Alcendor RR, Gao S, Zhai P et al (2007) Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res 100:1512–1521
Becatti M, Taddei N, Cecchi C et al (2012) SIRT1 modulates MAPK pathways in ischemic-reperfused cardiomyocytes. Cell Mol Life Sci 69:2245–2260
El-Mowafy AM, White RE (1999) Resveratrol inhibits MAPK activity and nuclear translocation in coronary artery smooth muscle: reversal of endothelin-1 stimulatory effects. FEBS Lett 451:63–67
El-Mowafy AM, Alkhalaf M, El-Kashef HA (2008) Resveratrol reverses hydrogen peroxide-induced proliferative effects in human coronary smooth muscle cells: a novel signaling mechanism. Arch Med Res 39:155–161
Haider UG, Sorescu D, Griendling KK et al (2002) Resveratrol suppresses angiotensin II-induced Akt/protein kinase B and p70 S6 kinase phosphorylation and subsequent hypertrophy in rat aortic smooth muscle cells. Mol Pharmacol 62:772–777
Lv XC, Zhou HY (2012) Resveratrol protects H9c2 embryonic rat heart derived cells from oxidative stress by inducing autophagy: role of p38 mitogen-activated protein kinase. Can J Physiol Pharmacol 90:655–662
Li H, Xia N, Forstermann U (2012) Cardiovascular effects and molecular targets of resveratrol. Nitric Oxide 26:102–110
Liu Z, Song Y, Zhang X et al (2005) Effects of trans-resveratrol on hypertension-induced cardiac hypertrophy using the partially nephrectomized rat model. Clin Exp Pharmacol Physiol 32:1049–1054
Toklu HZ, Sehirli O, Ersahin M et al (2010) Resveratrol improves cardiovascular function and reduces oxidative organ damage in the renal, cardiovascular and cerebral tissues of two-kidney, one-clip hypertensive rats. J Pharm Pharmacol 62:1784–1793
Chan V, Fenning A, Iyer A et al (2011) Resveratrol improves cardiovascular function in DOCA-salt hypertensive rats. Curr Pharm Biotechnol 12:429–436
Juric D, Wojciechowski P, Das DK et al (2007) Prevention of concentric hypertrophy and diastolic impairment in aortic-banded rats treated with resveratrol. Am J Physiol Heart Circ Physiol 292:H2138–H2143
Wang S, Wang X, Yan J et al (2007) Resveratrol inhibits proliferation of cultured rat cardiac fibroblasts: correlated with NO-cGMP signaling pathway. Eur J Pharmacol 567:26–35
Danz ED, Skramsted J, Henry N et al (2009) Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic Biol Med 46:1589–1597
Tatlidede E, Sehirli O, Velioglu-Ogunc A et al (2009) Resveratrol treatment protects against doxorubicin-induced cardiotoxicity by alleviating oxidative damage. Free Radic Res 43:195–205
Tanno M, Kuno A, Yano T et al (2010) Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J Biol Chem 285:8375–8382
Ghanim H, Sia CL, Korzeniewski K et al (2011) A resveratrol and polyphenol preparation suppresses oxidative and inflammatory stress response to a high-fat, high-carbohydrate meal. J Clin Endocrinol Metab 96:1409–1414
Magyar K, Halmosi R, Palfi A et al (2012) Cardioprotection by resveratrol: a human clinical trial in patients with stable coronary artery disease. Clin Hemorheol Microcirc 50:179–187
Frohlich ED, Gonzalez A, Diez J (2011) Hypertensive left ventricular hypertrophy risk: beyond adaptive cardiomyocytic hypertrophy. J Hypertens 29:17–26
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Hamza, S.M., Sung, M.M., Dyck, J.R.B. (2013). Reducing Oxidative Stress and Manipulating Molecular Signaling Events Using Resveratrol as a Therapy for Pathological Cardiac Hypertrophy. In: Jugdutt, B., Dhalla, N. (eds) Cardiac Remodeling. Advances in Biochemistry in Health and Disease, vol 5. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5930-9_14
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5930-9_14
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5929-3
Online ISBN: 978-1-4614-5930-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)