
Abstract
Over the course of evolution, aerobic organisms have developed sophisticated systems for responding to alterations in oxygen concentration, as oxygen acts as a final electron acceptor in oxidative phosphorylation for energy production. Hypoxia-inducible factor (HIF) plays a central role in the adaptive regulation of energy metabolism, by triggering a switch from mitochondrial oxidative phosphorylation to anaerobic glycolysis in hypoxic conditions. HIF also reduces oxygen consumption in mitochondria by inhibiting conversion of pyruvate to acetyl CoA, suppressing mitochondrial biogenesis and activating autophagy of mitochondria concomitantly with reduction in reactive oxygen species production. In addition, metabolic reprogramming in response to hypoxia through HIF activation is not limited to the regulation of carbohydrate metabolism; it occurs in lipid metabolism as well. Recent studies using in vivo gene-targeting technique have revealed unexpected, but novel functions of HIF in energy metabolism in a context- and cell type-specific manner, and shed light on the possibility of pharmaceutical targeting HIF as a new therapy against many diseases, including cancer, diabetes, and fatty liver.
Similar content being viewed by others
Introduction
Oxygen is an essential molecule for the survival of aerobic organisms, as it serves as the final electron acceptor in oxidative phosphorylation for energy production. Electron transfer of the respiratory chain in mitochondria is not completely efficient, and leakage of electrons occurs and produces reactive oxygen species (ROS), such as superoxide anion and hydrogen peroxide, by directly reducing oxygen. Although cells are equipped with anti-oxidant systems, unscavenged ROS have a highly oxidizing effect on cellular components, such as lipids, proteins, and nucleotides, which may threaten cell survival. Oxygen concentration must therefore be regulated tightly to balance oxygen demand and supply, by which cells can maintain ATP production with less ROS generation.
Hypoxia (lowered oxygen conditions) can be caused by not only reduced oxygen supply from local circulatory system (e.g. cancer, ischemia heart disease, and embryos), but also increased oxygen consumption by cells engaged in certain functions (e.g. inflammation, proliferation, and hormone secretion). Hypoxia-inducible factor (HIF) is a central transcription factor that enables adaptive response to hypoxic stress in normal and pathological conditions by activating a large number of genes responsible for oxygen delivery, angiogenesis, cell proliferation, cell differentiation, and metabolism [1, 2]. As energy production is linked intimately with almost all cellular events, we will describe first how HIFs play key roles in the regulation of cellular energy metabolism in response to hypoxia, and then discuss the complex range of functions of HIFs in whole-body energy metabolism in health and diseases.
HIF-1 determines fate of pyruvate to be metabolized to lactate in hypoxia
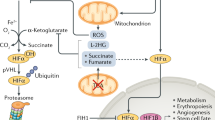
To adapt to oxygen deprivation, cells simultaneously undergo two dramatic alterations of cellular metabolism: oxygen-independent ATP production, and reduction of mitochondrial oxygen consumption (Fig. 1). The former “fermentative” energy production is under the control of HIF-1 activity, which stimulates glycolytic flux by inducing the expression of glucose transporters (GLUT1 and GLUT3) [3, 4] and glycolytic enzymes including hexokinase (HK1 and HK2) [5] and phosphoglycerate kinase 1 (PGK1) [6]. Although glycolysis produces less ATP per glucose molecule compared to oxidative phosphorylation, cooperative induction of glucose uptake and glycolysis can achieve rapid energy production that compensates for its low efficiency. In addition, HIF-1 also upregulates lactate dehydrogenase A (LDHA) [7], which regenerates NAD+ for continuous supply for glycolysis. The hypoxic alterations in metabolism are essential for hematopoietic stem cells to maintain their stemness in bone marrow niche, where oxygen concentrations are low enough to activate HIF-1 constitutively under physiological conditions [8, 9], and it has been reported that the Meis1-HIF-1 cascade is involved in enhanced glycolysis [10]. In contrast to normal cells, cancerous cells have long been known to show high glycolytic rates even under normoxic conditions (Warburg effect) [11]. Oncogenes, such as c-Myc and v-Src, have been shown to promote metabolic reprogramming, in part, by cooperation with and activation of HIF-1, respectively [12, 13]. In addition, loss of tumor suppressor genes, including PTEN [14] and VHL [15], has been implicated in development of the Warburg effects via HIF-1 activation, independent of oxygen concentration. While these reports have clearly shown the importance of HIF-1 in the maintenance of highly activated state of glycolysis in cancer cells, Luo et al. [16] have recently reported intricate interactions between glycolysis and HIF via PHD3. Pyruvate kinase M2 (PKM2) is an alternative form of PK expressed predominantly in embryonic and cancer cells, which catalyzes last step of irreversible reactions in glycolysis, and is induced by hypoxia in a HIF-1-dependent manner. Although hypoxic PKM2 induction itself can enhance HIF-1 transcriptional activity to some extent, hydroxylation of PKM2 by PHD3 is a prerequisite for the full function of HIF-1 in hypoxia, and thus the maintenance of highly activated glycolysis, providing a positive feedback loop to reprogram a characteristic glucose metabolism in cancer cells.

HIF-1-dependent regulation of carbohydrate and energy metabolism. Under hypoxia, HIF-1 activates glucose intake, glycolysis, and conversion of pyruvate to lactate, by which ATP production is maintained even in oxygen deprivation. On the other hand, HIF also reduces mitochondrial oxygen consumption by inhibiting mitochondrial biogenesis and activating mitophagy. Molecules activated by HIF-1 is drawn in red box
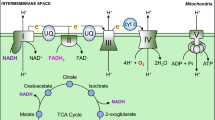
Reduction of oxygen consumption in mitochondria serves as a safeguard for cell survival under hypoxia by inhibiting aberrant electron leakages from mitochondria and, in turn, preventing ROS production. As NADH, which is abundantly produced in the TCA cycle, stimulates mitochondrial oxygen consumption, entry of acetyl CoA into TCA cycle must be diminished under hypoxia. Acetyl CoA is produced only by oxidative decarboxylation of pyruvate or β-oxidation of fatty acids. Pyruvate dehydrogenase (PDH) is an E1 component of a large multienzyme complex responsible for conversion of pyruvate to acetyl CoA, and its enzymatic activity is inactivated substantially by phosphorylation at specific serine residues by four PDH kinases (PDKs). Of these, PDK1 and -3 are well-known targets of HIF-1 [17, 18], and PDK1 can phosphorylate all three sites of serine residues in PDH, conferring extreme resistance to dephosphorylation (reactivation) to the enzyme [19]. Upon exposure to hypoxia, HIF-1-mediated PDK1 induction in conjunction with LDHA shunts pyruvate away from the mitochondria, which reduces flux through TCA cycle and delivery of NADH to mitochondrial respiratory chain. In addition, hypoxia-elicited PDH inhibition by PDK1 is not likely to be abrogated immediately after reoxygenation, compared to such inhibition by other PDKs as seen in starvation, thus providing a protective mechanism against reoxygenation-associated mitochondrial ROS production by slowing restoration of acetyl CoA entry via PDH into TCA cycle. In agreement with these notions, mouse embryonic fibroblasts (MEFs) lacking HIF-1α gene undergo cell death as a result of excess ROS production due to a failure of PDK1 induction [20]. A recent report by Aragones et al. [21] shows critical roles of PDK4 in a shift in glucose metabolism from inside to outside of the mitochondria in PHD1-null muscle, and such an induction is attributable to an activation of PPARα, in part, by HIF-2. Besides PDK induction, reduced activity in TCA cycle enzyme(s) also may present other regulatory mechanism to control mitochondrial oxygen consumption. In response to hypoxia, HIF-1 has recently been reported to induce transcription of a microRNA, miR-210, which decreases expression of iron-sulfur scaffold proteins ISCU1/2, essential components for assembly of iron–sulfur cluster [22]. As iron–sulfur cluster is a prerequisite for the activity of aconitase, subunit D of succinate dehydrogenase, complex I, and COX subunit10, increased miR-210 inhibits TCA cycle and oxidative phosphorylation, leading to a reduction in mitochondrial oxygen consumption under hypoxia [23].
Hypoxic stress reduces oxygen consumption by remodeling mitochondria
Alternative metabolic reprogramming that leads to decreased oxygen consumption causes drastic organelle remodeling in mitochondria (Fig. 1). In VHL-deficient renal carcinoma cells, HIF-1 blocks mitochondrial biogenesis by inducing MXI-1, which interferes with c-Myc-Max interaction and consequently represses transcriptional activity of PGC-1α, a critical transcription factor for mitochondrial biogenesis [24]. Recent report by Jensen et al. [25] suggests that HIF-1-mediated FoxO3A directly antagonizes c-Myc function, resulting in hypoxic suppression of mitochondrial mass and oxygen consumption with metabolic shifts to glycolysis. It has also been reported that HIF-1-mediated miR-210 induction activates another c-Myc antagonist, MNT, and may be involved in the regulation of mitochondrial mass through competition with Max for c-Myc binding in a similar way of MXI-1 [26]. On the other hand, HIF-2 also counteracts c-Myc activity and inhibits mitochondrial biogenesis in a manner similar to HIF-1 [24]. However, controversy remains regarding the role of HIF-2 in the regulation of mitochondrial mass, as Gordan and colleagues [27] have reported that HIF-2α conversely enhances c-Myc transcriptional activity by binding Max, resulting in the promotion of renal cell carcinoma. A second key remodeling of mitochondria is evoked by the induction of BNIP3, a member of BCL2 family. HIF-1-mediated BNIP3 expression impedes association of Bcl-2/xL and Beclin-1 (also known as atg6, an essential component of autophagy), and then released Beclin-1 promotes selective autophagy of mitochondria (mitophagy), resulting in mitochondrial mass reduction [28]. In fact, HIF-1α-deficient MEFs fails to decrease mitochondrial mass and oxygen consumption due to a lack of BNIP3 induction, leading to increase in ROS production and cell death under hypoxia [28]. In addition, BNIP3 may participate in hypoxia-induced mitochondrial fission by inhibiting a mitochondrial fusion-associated protein, OPA1, thereby accelerating mitophagy of dysfunctional mitochondria, a putative source of ROS production [29].
Although reduction in mitochondrial mass is the most effective way to limit oxygen consumption in hypoxia, mitochondrial respiration is not halted completely unless oxygen tension drops to extreme low levels (<0.3 % oxygen), suggesting that mitochondria still functions as ATP generating system in hypoxic conditions. This idea is supported by the fact that HIF-1 regulates COX activity by switching from COX4-1 to COX4-2 [30]. This subunit conversion is also promoted by progressive degradation of COX4-1 by LON, which operates as a mitochondrial protease under hypoxia, and is upregulated by HIF-1 [30]. Thus, the subunit switch serves as a promising metabolic response to ensure efficient ATP production with lower ROS generation in hypoxic cells.
In vivo functions of HIF-1 on carbohydrate metabolism
Emerging data on metabolic adaptation to hypoxia have stemmed largely from works using cancer cells, suggesting the importance of HIF-1 in energy homeostasis under low oxygen conditions. Next, we will discuss roles of HIF in the regulation of whole-body energy metabolism, focusing particularly on the liver, a central organ of energy metabolism. As inactivation of HIFαs and HIF-related gene results in embryonic lethality [31], a conditional targeting of the gene has been conducted to investigate in vivo function of HIFs. Surprisingly, Kahn and colleagues showed that deletion of HIF-1β gene in liver causes impaired glucose tolerance with normal fasting blood glucose levels only in male mice, and the diabetic phenotype is attributable, in part, to aberrant activation of C/EBPα and enhanced gluconeogenesis [32]. In addition, liver-specific VHL-deficient mice show great reduction in the expression of gluconeogenetic enzymes and, thus, suffer from severe hypoglycemia, which are accompanied by decreased levels in HNF4α and PGC1α [33]. These data suggest that basal levels of HIFαs expression are a prerequisite for suppression of hepatic gluconeogenesis through complicated interactions with other transcription factors. On the other hand, loss of HIF-1α has no substantial effect on basal carbohydrate metabolism and the related gene expressions, including GLUT1, GK, and PGK1 in the liver, although isolated primary hepatocytes can respond to hypoxia by enhancing GK and PGK1 expression (unpublished data). These data suggest that a compensatory safeguard system mediated by HIF-2 or other transcription factors may operate in vivo.
We have next investigated role of HIF-1 in liver regeneration, during which liver gluconeogenesis and glycolysis are, respectively, highly activated and suppressed to maintain whole-body energy metabolism. It is worth noting that hypoxia-induced HIF-1 target genes, such as PKL and inducible PFK, are not altered in regenerated liver, although HIF-1α activity is transiently induced, presumably by decreased liver perfusion in liver regeneration [34]. Unexpectedly, hepatic gluconeogenesis is markedly suppressed through a failure of induction of PEPCK and PGK1 in HIF-1α-deficient mice, resulting in severe fasting hypoglycemia during liver regeneration. These data suggest that HIF-1 induction is necessary for the regenerated liver to activate gluconeogenesis, rather than glycolysis, to maintain whole-body energy metabolism. In addition, these mice accumulate liver glycogen in the fed state to greater extents compared to control mice presumably by an aberrant activation of AKT-GSK3β signaling. While these findings appear to conflict with a previous report that showed hypoxic glycogen accumulation, the later response seen in a certain cell line results from HIF-1-mediated activation of the muscle type of glycogen synthase (GYS1), but not the liver one (GYS2) [35], suggesting a tissue-specific regulation of glycogen metabolism in response to HIF-1 activation. In diet-induced obese mice, we have found that HIF-1α expression is also transiently induced with expansion of the hypoxic area in the affected liver [36]. Mice lacking functional HIF-1α gene show severe glucose intolerance by impaired β cell functions and insulin resistance in peripheral tissues such as skeletal muscles and adipose tissues after long-term treatments with high-fat/sucrose diets [36]. The metabolic alterations seen in HIF-1α-deficient mice appear to be secondary to impaired hepatic glucokinase (GK) induction and the resultant reduction in postprandial glucose uptake by the liver, such that forced GK induction restores the capacity for hepatic glucose disposal. However, GLUT1 and other glycolytic enzymes such as PGK1 are expressed comparably between control and HIF-1α-deficient mice fed the diet. Collectively, although HIF-1α expression is detectable under physiological conditions and induced in certain types of diseases in the liver, many hypoxic changes of genes involved in the energy homeostasis seen in cell lines (e.g. activation of glycolysis) are not reproduced in the liver, strongly suggesting that HIF-1-dependent metabolic shift occurs in a context- and tissue-dependent manner. In conclusion, the targeting of HIF-1 to modulate hepatic glucose metabolism may provide a promising therapy against diabetes and postsurgical liver dysfunction, but additional studies are needed to examine in detail what affects HIF-1-mediated regulations of carbohydrate metabolism in the liver.
Complicated HIFs regulation of lipid metabolism
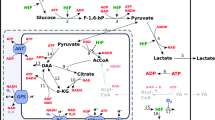
As oxidative breakdown of fatty acids consumes a large amount of oxygen to produce energy, it is conceivable that lipid metabolism should be regulated through activation of HIFs in hypoxia. It has been reported that, in certain cell types, the expression of PPARα and/or RXR, an obligate binding partner of PPARα, or the ability of DNA-binding affinity of PPARα/RXR complex are reduced under hypoxia, in part, in a HIF-1-dependent manner [37]. In contrast, liver-specific VHL-deficient mice disclose the importance of HIF-2 rather than HIF-1 in the regulation of β oxidation and lipid droplet formation, by which a constitutive activation of HIF-2α causes severe lipid accumulation in the VHL-deficient liver [38]. This is further supported by a recent study showing development of severe fatty liver in PHD2/3 double-knockout mice in a HIF-2-dependent manner [39]. However, much controversy remains regarding the roles of HIF-2 as a pro-lipogenic factor, inasmuch as HIF-2α-deficient mice also exhibit hepatic steatosis [40] and forced expression of HIF-1α, but not HIF-2α, in liver stimulates lipid accumulation in mice [41]. Moreover, HIF-1 has been reported to induce HIG2 expression, a lipid droplet protein involved in neutral lipid formation, under hypoxia, [42] and accelerate fatty acid synthesis through simultaneous activations of PPARγ and glycolysis in cardiac hypertrophy [43].
Although the data described above suggest that HIF activation results in lipid accumulation by several mechanisms, we have recently reported a converse function of HIF-1 in lipid metabolism in alcoholic fatty liver. Chronic intakes of excessive alcohol have been known to accumulate neutral lipids in liver and cause liver hypoxia concomitantly with increased oxygen consumption in the affected liver. In fact, such a disrupted oxygen homeostasis induces hepatic HIF-1α expression and its transcriptional activity in liver of mice exposed to alcohol only for 4 weeks [44]. Loss of HIF-1α deteriorates fatty infiltration predominantly in the pericentral region of the liver compared to control mice, suggesting anti-lipogenic roles of HIF-1 in alcoholic fatty liver. In the mutant mice, enhanced hepatic lipid accumulation is accompanied by aberrant induction of SREBP1c and ACC1, and these transcriptional alterations are evoked by a lack of HIF-1-mediated DEC1 activation. This idea is supported by the finding that forced expression of a transcriptional repressor, DEC1, in the mutant liver completely suppresses lipid accumulation by reducing SREBP1c activation. In addition, a PHD inhibitor dimethyloxalylglycine, which stabilize and activate HIFα, reduces alcoholic-elicited lipid deposition in control, but not HIF-1α-deficient mice, confirming the protective roles of HIF-1 against alcoholic fatty liver [44]. Consistent with these findings, deletion of the HIF-2α gene in liver does not show any effects on lipid accumulation in the same disease model (unpublished data). Collectively, these data strongly suggest that HIF-1 protects against development of alcoholic fatty liver by inhibiting de novo fatty acid synthesis. More importantly for liver biology, our study appears to raise complex, but important issues for the understanding of in vivo functions of HIFs in the regulation of liver metabolism, as hepatocytes show distinct metabolic properties depending on their locations in the liver acinus. In lipid metabolism, lipogenesis and fatty acid oxidation occur predominantly in pericentral (less-oxygenated) and periportal (well-oxygenated) region of liver acinus [45]. When HIFα expression is forced by either inactivation of VHL gene or adenovirus vector encoding HIFα gene, HIF-2 may regulate genes responsible for fatty acid oxidation, rather than those for de novo lipogenesis, as ubiquitous activation of HIFαs can occur in the acinus. Selective functions of HIF-1 (but not HIF-2) on fatty acid synthesis could alternatively be at work, even if both HIFαs are activated exclusively in pericentral hepatocytes. In either case, the HIF-mediated metabolic alterations described above appears important for the maintenance of local oxygen homeostasis in the liver by limiting either oxygen consumption (fatty acid oxidation) or ATP utilization (de novo fatty acid synthesis), which are characteristic responses in cells evoked by hypoxia (Fig. 2).

HIFs-mediated lipid metabolism in liver. HIF-1 and HIF-2 inhibits de novo fatty acids synthesis and β-oxidation, respectively, in the liver. In addition, HIF-2 also stimulates lipid droplet formation to promote triacylglycerol storage. Although HIF-mediated regulation of lipid metabolism is complicated, HIF activation appears to lead to reduction in consumption of both ATP and oxygen
Conclusion
Metabolic reprogramming to hypoxia mediated by HIFs is critically important for aerobic cells (organisms) to survive as oxygen concentrations surrounding them vary drastically in disease conditions, and even under physiological ones. Much information as to how HIFs are activated and what genes are regulated by HIFs is now available to easily understand the importance of HIFs in hypoxic adaptation. Although HIF-1 and HIF-2 can be activated simultaneously or separately by hypoxia (and certain types of human diseases) in one cell, the consequences on cellular and tissue metabolism appear to differ markedly in a cell type- and context-dependent manner. Further investigation of the roles of HIFs in different cells and conditions is needed to gain a better understanding of the whole picture of their physiological and pathophysiological roles, and is essential for developing novel therapeutic strategies against diseases.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- BCL2:
-
B-cell lymphoma 2
- BNIP3:
-
BCL2/adenovirus E1B 19 kd-interacting protein
- C/EBP:
-
CCAAT/enhancer binding protein
- COX:
-
Cytochrome c oxidase
- DEC:
-
Differentiated embryo chondrocyte
- GK:
-
Glucokinase
- GLUT:
-
Glucose transporters
- HIF:
-
Hypoxia inducible factor
- HK:
-
Hexokinase
- HNF:
-
Hepatocyte nuclear factor
- LDH:
-
Lactate dehydrogenase
- MEF:
-
Mouse embryonic fibroblast
- NADH:
-
Nicotinamide adenine dinucleotide
- PDH:
-
Pyruvate dehydrogenase
- PDK:
-
PDH kinase
- PGC:
-
PPAR-γ coactivator, PGC
- PGK:
-
Phosphoglycerate kinase
- PHD:
-
Prolyl hydroxylase domain
- PK:
-
Pyruvate kinase
- PPAR:
-
Peroxisome proliferator-activated receptor
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- PTEN:
-
Phosphatase and tensin homolog deleted from chromosome 10
- RXR:
-
Retinoid X receptor
- ROS:
-
Reactive oxygen species
- SREBP:
-
Sterol regulatory element-binding protein
- TCA:
-
Tricarboxylic acid
- VHL:
-
von Hipple-Lindau
References
Poellinger L, Johnson RS. HIF-1 and hypoxic response: the plot thickens. Curr Opin Genet Dev. 2004;14:81–5.
Semenza GL. Life with oxygen. Science. 2007;318:62–4.
Gleadle JM, Ratcliffe PJ. Induction of hypoxia-inducible factor-1, erythropoietin, vascular endothelial growth factor, and glucose transporter-1 by hypoxia: evidence against a regulatory role for Src kinase. Blood. 1997;89:503–9.
Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA. 1997;94:8104–9.
Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem. 2001;276:43407–12.
Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269:23757–63.
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–37.
Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9:298–310.
Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402.
Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–90.
Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–30.
Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27:7381–93.
Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 1997;57:5328–35.
Tandon P, Gallo CA, Khatri S, Barger JF, Yepiskoposyan H, Plas DR. Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency. Proc Natl Acad Sci USA. 2011;108:2361–5.
Minchenko A, Leshchinsky I, Opentanova I, Sang N, Srinivas V, Armstead V, et al. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J Biol Chem. 2002;277:6183–7.
Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–44.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85.
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–97.
Sugden MC, Holness MJ. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem. 2006;112:139–49.
Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. 2008;15:621–7.
Aragones J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40:170–80.
Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10:273–84.
Chen Z, Li Y, Zhang H, Huang P, Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. 2010;29:4362–8.
Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–20.
Jensen KS, Binderup T, Jensen KT, Therkelsen I, Borup R, Nilsson E, et al. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 2011;30:4554–70.
Zhang Z, Sun H, Dai H, Walsh RM, Imakura M, Schelter J, et al. MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle. 2009;8:2756–68.
Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903.
Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnaune-Pelloquin L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010;11:459–65.
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–22.
Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda). 2009;24:97–106.
Wang XL, Suzuki R, Lee K, Tran T, Gunton JE, Saha AK, et al. Ablation of ARNT/HIF1beta in liver alters gluconeogenesis, lipogenic gene expression, and serum ketones. Cell Metab. 2009;9:428–39.
Kucejova B, Sunny NE, Nguyen AD, Hallac R, Fu X, Pena-Llopis S, et al. Uncoupling hypoxia signaling from oxygen sensing in the liver results in hypoketotic hypoglycemic death. Oncogene. 2011;30:2147–60.
Tajima T, Goda N, Fujiki N, Hishiki T, Nishiyama Y, Senoo-Matsuda N, et al. HIF-1alpha is necessary to support gluconeogenesis during liver regeneration. Biochem Biophys Res Commun. 2009;387:789–94.
Pescador N, Villar D, Cifuentes D, Garcia-Rocha M, Ortiz-Barahona A, Vazquez S, et al. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS ONE. 2010;5:e9644.
Ochiai D, Goda N, Hishiki T, Kanai M, Senoo-Matsuda N, Soga T, et al. Disruption of HIF-1alpha in hepatocytes impairs glucose metabolism in diet-induced obesity mice. Biochem Biophys Res Commun. 2011;415:445–9.
Narravula S, Colgan SP. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol. 2001;166:7543–8.
Rankin EB, Rha J, Selak MA, Unger TL, Keith B, Liu Q, et al. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol Cell Biol. 2009;29:4527–38.
Minamishima YA, Moslehi J, Padera RF, Bronson RT, Liao R, Kaelin WG Jr. A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response in vivo. Mol Cell Biol. 2009;29:5729–41.
Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat Genet. 2003;35:331–40.
Kim WY, Safran M, Buckley MR, Ebert BL, Glickman J, Bosenberg M, et al. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J. 2006;25:4650–62.
Gimm T, Wiese M, Teschemacher B, Deggerich A, Schodel J, Knaup KX, et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010;24:4443–58.
Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9:512–24.
Nishiyama Y, Goda N, Kanai M, Niwa D, Osanai K, Yamamoto Y, et al. HIF-1alpha induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J Hepatol. 2012;56:441–7.
Guzman M, Castro J. Zonation of fatty acid metabolism in rat liver. Biochem J. 1989;264:107–13.
Acknowledgments
This work was supported, in part, by a grant from The NOVARTIS Foundation (Japan) for the Promotion of Science, a grant from ASAHI BREWERIES Foundation, a grant from Waseda University Grant For Special Research Projects, a grant from the “High-Tech Research Center” Project for Private Universities, with a matching fund subsidy from MEXT, and a Grant-in-Aid for Scientific Research (C) from MEXT.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Goda, N., Kanai, M. Hypoxia-inducible factors and their roles in energy metabolism. Int J Hematol 95, 457–463 (2012). https://doi.org/10.1007/s12185-012-1069-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-012-1069-y