
Abstract
C-type lectin domain family 4, member M (CLEC4M), a trans-membrane protein specifically expressed in liver sinusoidal endothelial cells, is considered a candidate receptor for hepatotropism of hepatitis C virus (HCV). CLEC4M was previously reported to capture artificial HCVpp (pseudoparticle) and transmit it to hepatocytes (transinfection) via CLEC4M-positive cells. It is still not known whether CLEC4M acts as a receptor for HCVcc (cell-culture-produced HCV) transinfection or whether CLEC4M is an entry receptor for HCVcc. Initially, we established stably CLEC4M-positive and HCV-replication-permissive cell lines by introducing a CLEC4M expression vector into Huh7-25 cells (Huh7-25-CLEC4M) by transfection. Huh7-25 is a mutant cell line that is resistant to JFH-1 HCVcc due to the lack of expression of CD81 but permissive for replication of JFH1 HCV RNA. When Huh7-25-CLEC4M cells were infected with HCVcc and cultured for 6 days, none were positive for infection. Next, to examine whether CLEC4M functions as a receptor for transinfection, Huh7-25-CLEC4M cells were inoculated with HCVcc and thereafter co-cultured with Huh7-it cells, which are susceptible to HCV infection. The amount of HCV RNA was increased in Huh7-it cells co-cultured with Huh7-25-CLEC4M cells, and the transinfection was inhibited in the presence of anti-CLEC4M antibody during inoculation. Thus, CLEC4M cannot substitute for CD81 as an entry receptor for JFH-1 HCVcc. It just mediates transinfection without internalization of HCVcc. CD81 is still crucial for HCV entry into hepatocytes, and CLEC4M in liver sinusoidal endothelial cells may be responsible for hepatotropism of HCV infection by trapping circulating HCV to transmit it to adjacent hepatocytes.
Similar content being viewed by others

Introduction
Hepatitis C virus (HCV) causes serious liver diseases including chronic hepatitis, liver cirrhosis and hepatocellular carcinoma. HCV infection is manifested mainly in human liver hepatocytes [1] and hematopoietic cells [18]. Such strict tropism of HCV infection depends on two major factors: the ability of HCV to enter a target cell and the permissibility of HCV replication inside the cell. Target cell accessibility is mediated by HCV receptors and co-receptors, which have been identified using HCV cell culture systems with CD81, scavenger receptor class B, member 1 (SR-BI), low-density lipoprotein receptor (LDLR), claudin-1 (CLDN1), and occludin (OCLN) [7, 22]. Although CD81 and OCLN are essential determinants of tropism of HCV infection in humans [19], these cellular proteins are ubiquitously expressed in human tissues and are not responsible for the hepatotropism of HCV infection. One liver-specific molecule that may influence the accessibility of cells to pathogens is C-type lectin domain family 4, member M (CLEC4M; also known as L-SIGN or DC-SIGNR). CLEC4M is highly expressed on liver sinusoidal endothelial cells but not on hepatocytes and dendritic cells. [5]. Soilleux and colleagues identified the cDNA sequence of CLEC4M as a gene related to that of dendritic-cell-specific ICAM-3-grabbing non-integrin (DC-SIGN) [4]. The C-terminal end of CLEC4M possesses a carbohydrate-recognition domain and binds to mannose oligosaccharides that are present on the envelopes of pathogens [13]. CLEC4M acts as a capture receptor for viruses and pathogens, including human immunodeficiency virus type 1 (HIV-1) [5], Ebola virus [3], severe acute respiratory syndrome coronavirus (SARS-CoV) [8] and M tuberculosis [14]. CLEC4M was previously reported to mediate transinfection of HCVpp (pseudoparticle) using CLEC4M-positive cells [9, 10, 15, 16]; CLEC4M captured artificial HCVpp and transmitted it to hepatocytes. It is, however, unknown whether CLEC4M acts as a receptor for HCVcc (cell-culture-produced HCV) transinfection and whether CLEC4M is a binding receptor or an entry receptor for HCVcc. Once cells that are competent for HCV RNA replication have an entry receptor for HCVcc, they are susceptible to HCVcc infection, but cells that have only a binding receptor are unsusceptible. In the present study, we used CD81-negative and HCV-replication-permissive cells (Huh7-25) to investigate whether CLEC4M plays a role as a liver-specific HCV receptor for cisinfection and/or a receptor for transinfection.
Materials and methods
Cells
Huh7-25, Huh7-25-CD81 [2] and Huh7-it cells (a gift from Dr. Oshima, National Institute of Infection Diseases, Tokyo, Japan) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10 % fetal bovine serum. Huh7-25-CD81 cells were maintained in the complete medium containing 400 μg G418 (Invitrogen, Carlsbad, CA) per ml. Huh7-25-CD81 and Huh7-it cells were highly susceptible to HCVcc infection. Huh7-it cells were generated by Dr. Oshima from HCV replicon cells and cured by interferon treatment.
Transfection with CLEC4M expressing plasmids
Human CLEC4M-cDNA (isoform 1, accession no. NM_014257) was amplified from human liver cDNA using the primers shown in Supplementary Table 1. The amplified cDNA was inserted into the pEF1/V5-His vector containing a Geneticin-resistance gene (Invitrogen). The nucleotide sequence of the CLEC4M construct was confirmed to be correct. Huh7-25 cells were transfected with a complex of plasmid DNA and Lipofectamine LTX (Invitrogen), and stable clones of transfected cells were established by exposure to 400 μg of G418 (Invitrogen) per ml. Stable clones transfected with pEF1/V5-His vector were used as controls.
Quantification of mRNA
Total RNA was extracted from cultured cells using TRIzol Reagent (Invitrogen) and subjected to DNase I treatment followed by cDNA synthesis as described previously [12]. The quantity of mRNA was determined by real-time PCR using SYBR Green [12]. Plasmid DNA containing a target gene was used as a standard for quantification of HCV RNA and CLEC4M mRNA. CLEC4M gene expression was normalized against the quantity of GAPDH, and the relative value was expressed in arbitrary units. Primer sequences were designed using Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) and are shown in Supplementary Table 1.
Immunofluorescence staining
Cells seeded on cover slips were incubated overnight at 37 °C and fixed with cold acetone for 3 min. After fixation, the cells were blocked with 1 % bovine serum albumin in phosphate-buffered saline (PBS) and incubated with the first antibody: 5 μg/ml of mouse anti-CLEC4M antibody (mAb162) (R &D systems, Inc., Minneapolis, MN) and/or 200-fold diluted human serum from an HCV-positive donor. The human anti-HCV antiserum used was characterized and found to be reactive for core, NS4B and NS5A proteins of JFH-1 HCVcc by Western blotting of the lysate from infected cells (Supplementary Fig. 1). The cells were then stained with 4 μg/ml of the secondary antibody conjugated with AlexaFluor 488 or AlexaFluor 647 (Invitrogen). The stained cells were mounted on glass slides with ProLong Gold antifade reagent containing DAPI (Invitrogen) and visualized using a BIOREVO BZ-9000 microscope (KEYENCE, Osaka, Japan), which was also used to measure the fluorescence intensity of CLEC4M proteins.
Western blotting
Cells and human liver tissues were lysed using urea buffer (7 M urea, 2 M thiourea, 3 % CHAPS, 3 % Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 30 mM Tris-HCl, pH 7.4). Protein (50 μg) was treated with N-glycanase (PNGase F) included in the Enzymatic Protein Deglycosylation Kit (Sigma-Aldrich, Saint Louis, MO). Treated and untreated proteins (20-μg samples) were electrophoresed in an SDS-10 % polyacrylamide gel and then transferred to a nitrocellulose membrane. CLEC4M protein was detected using 0.5 μg of goat anti-CLEC4M antibody N17 (Santa Cruz Biotechnology. Inc., Santa Cruz, CA) and 7.5 ng of horseradish-peroxidase-conjugated anti-goat IgG (Zymed Laboratories, San Francisco, CA) per ml. β-actin protein, used as an internal loading standard, was detected using 1 μg of mouse monoclonal anti-β-actin antibody (Abcam, Cambridge, UK) and 0.1 μg of horseradish-peroxidase-conjugated anti-mouse IgG (IBL, Gunma, Japan) per ml. CLEC4M and β-actin were detected using ECL Advance and Plus Western Blotting Detection Systems (GE Healthcare, Buckinghamshire, UK), respectively. Chemiluminescence was detected with a Light-Capture AE-6972 (ATTO, Tokyo, Japan) and analyzed using CS Analyzer version 2.07 software (ATTO).
Reporter assay for HCV replication
Reporter replicon RNA was transcribed in vitro from plasmid pSGR-JFH1/Luc and introduced into Huh7 cells by transfection as described previously [2]. Luciferase activity was determined by using Luciferase Assay Systems (Promega, Madison, WI) and an Infinite F200 microplate reader (TECAN, Männedorf, Switzerland).
Production of infectious HCVcc
HCVcc derived from JFH-1 was prepared as described previously [20]. To produce a large amount of HCVcc, 3.6 × 106 Huh7-it cells were inoculated with 500 μl of HCVcc (8 × 106 HCV RNA copies/ml) for 1 h and cultured with 15 ml of the complete medium. The culture supernatant was collected at 6 days postinfection and passed through a 0.45-μm filter. Filtered supernatant was stored at −80 °C until use. The infectious titer of JFH-1 HCVcc was 9.2 × 103 focus-forming units (ffu) per ml, and the RNA titer was 2.0 × 107 copies of HCV RNA/ml.
Cisinfection with HCVcc
Huh7-25-CLEC4M cells (2 × 104) were seeded on coverslips in 24-well plates and incubated overnight at 37 °C. The cells were incubated with 200 μl of HCVcc for 4 h and then cultured in 500 μl of complete medium. At 6 days postinfection, infected cells were detected by immunofluorescence staining of HCV proteins. The infected foci were counted using fluorescence microscopy (Olympus, Tokyo, Japan). The assay was performed in triplicate.
Transinfection with HCVcc
Huh7-25-CLEC4M cells were seeded at 1 × 104 cells per well in 24-well plates. The cells were inoculated 24 h later with 200 μl of HCVcc for 4 h, and then washed five times in wash buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM CaCl2, 2 mM MgCl2, 0.5 % bovine serum albumin). After washing to remove unbound HCVcc, Huh7-it cells (4 × 104 cells/500 μl of the complete medium) were added to the cells and co-cultured for 4 h or 4 days. Then, total RNA was extracted from the co-cultured cells and the quantity of HCV RNA was determined. The co-cultured cells on cover slips were also stained for CLEC4M and HCV proteins by immunofluorescence staining. In the transinfection inhibition assay, Huh7-25-CLEC4M cells were preincubated in 200 μl of complete medium containing MAb162 for 30 min at 37 °C, followed by infection with HCVcc. MAb162 specifically recognizes the carbohydrate-recognition domain of CLEC4M. The assay was performed in triplicate.
Statistical analysis
Student’s t-test and the Mann-Whitney U test were used to analyze the data. P-values less than 0.05 were considered statistically significant.
Results
Characterization of CLEC4M-positive cells
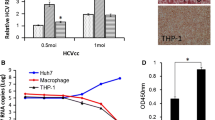
To investigate the potential receptor role of CLEC4M in HCV infection, we produced CLEC4M-positive cells using a CD81-negative Huh7 cell clone (Huh7-25). The Huh7-25 cell clone is highly permissive for HCV replication, but it is not susceptible to JFH-1 HCV infection because the Huh7-25 cell clone is negative for CD81 [2]. However, except for CD81, the expression level of HCV receptor genes in the Huh7-25 cell clone was similar to that of JFH-1 HCV permissive cells (Huh7-25-CD81 and Huh7-it) (Supplementary Fig. 2). Since Huh7-25 was also negative for CLEC4M mRNA, we established CLEC4M-positive cells using Huh7-25 (Huh7-25-CLEC4M). Huh7-25-CLEC4M cells produced high levels of CLEC4M mRNA and protein (Fig. 1A-C). Western blot analysis of the CLEC4M protein showed multiple bands of different sizes at >43 kDa in both Huh7-25-CLEC4M cells and human liver. These were converted to a single band of 43 kDa after treatment with N-glycanase (Fig. 1D). Thus, the glycosylation pattern of CLEC4M was different in Huh7-25-CLEC4M cells and human liver. Huh7-25-CLEC4M was similar to not only the negative control cell clone (Huh7-25-NC) but also to Huh7-25-CD81 cells, which were established by transfecting Huh7-25 cells with CD81, transforming them into HCV-susceptible cells [2] (Fig. 1E). Thus, the Huh7-25-CLEC4M cell clone was useful for determining whether CLEC4M is an entry receptor for HCVcc that can take the place of CD81.

Characterization of Huh7-25-CLEC4M cells. (A) Quantitation of CLEC4M mRNA in Huh7-25-CLEC4M cells and human liver tissue (HL). The mRNA levels were quantified by real-time PCR. Gene expression was normalized to the quantity of GAPDH used as an internal control gene, and the relative value was expressed in arbitrary units. (B) Immunofluorescence staining of CLEC4M. Huh7-25-CLEC4M cells were stained for CLEC4M by immunofluorescence (green) and for nuclei with DAPI (blue). CLEC4M on the cell surface was detected by binding of monoclonal anti-CLEC4M antibody before fixation with cold acetone. Huh7-25-NC cells were transfected with the pEF1/V5-His plasmid. Liver sinusoidal endothelial cells of HL were also positive for CLEC4M. Bar, 100 μm. (C) Fluorescence intensity of CLEC4M protein in (B). Fluorescence intensity was calculated as the ratio of total CLEC4M fluorescence to the total area of the cells examined. (D) Western blotting of CLEC4M in Huh7-25-CLEC4M cells and HL. Several bands around 50 kDa and a single 43-kDa band were specific for CLEC4M in untreated and deglycosylated samples, respectively. (E) Luciferase reporter assay of HCV replication in Huh7-25-CLEC4M and -NC cells. Luciferase activity was measured 4 and 48 h after transfection with pSGR-JFH1/Luc, and the relative luciferase activity was expressed as the ratio between the two values. HCV-susceptible Huh7-25-CD81 cells were used as a positive control. The assay was performed in duplicate
No cisinfection of Huh7-25-CLEC4M cells with HCVcc
To examine whether CLEC4M acts as a receptor for cisinfection, Huh7-25-CLEC4M cells were inoculated with HCVcc (1.8 × 103 ffu). Six days after HCV infection, none of the Huh7-25-CLEC4M or -NC cells were positive for HCV. In contrast, 70-80 % of Huh7-25-CD81 cells were positive for HCV (Fig. 2). Thus, CLEC4M is not an entry receptor for HCV, nor does it substitute for CD81 to mediate HCV cisinfection.

Infectivity in Hu7-25-CLEC4M cells. Hu7-25-CLEC4M, -NC, and -CD81 cells were stained for HCV proteins by immunofluorescence (green) and for nuclei with DAPI (blue) 6 days postinfection. Huh7-25-CD81 cells were used as a positive control. The assay was performed in triplicate
Transinfection of Huh7-25-CLEC4M cells with HCVcc
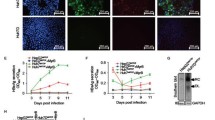
Next, we investigated whether CLEC4M is involved in transinfection with HCVcc. Four hours after inoculation with HCVcc, the amount of HCV RNA in Huh7-25-CLEC4M cells was the same as in Huh7-25-NC cells (Fig. 3A). The amount of attachment of HCVcc was similar with Huh7-25-CLEC4M and -NC cells. However, the level of HCV RNA after 4 days of cultivation increased 3-fold in Huh7-it cells co-cultured with Huh7-25-CLEC4M cells, and 13-fold in comparison to the negative control (Fig. 3A). As shown by immunofluorescence staining of HCV and CLEC4M after 3 days of co-culture, HCV-positive Huh7-it cells were adjacent to the Huh7-25-CLEC4M cells (Fig. 3B). To confirm CLEC4M-dependent transinfection, Huh7-25-CLEC4M cells were treated with anti-CLEC4M antibody before inoculation with HCVcc. The amount of HCV RNA was decreased specifically by treatment with anti-CLEC4M antibody (Fig. 4), but not in the case of Huh7-it cells (CD81-positive and CLEC4M-negative cells). These results suggest that CLEC4M is a receptor for HCV transinfection.

CLEC4M-mediated transinfection of Huh7-it cells with JFH-1 HCVcc. (A) Quantitation of HCV RNA 4 h (day 0) and 4 days (day 4) postinfection. Hu7-25-CLEC4M and -NC cells were incubated with JFH-1 HCVcc for 4 h at 37 °C. After being washed, cells were co-cultured with Huh7-it cells. The cells were harvested 4 days later, and HCV RNA was measured by real-time PCR. Bars indicate standard deviation of triplicate experiments. (B) Immunofluorescence staining of CLEC4M and HCV protein. Co-cultured cells were stained for CLEC4M (green) and HCV protein (red) by immunofluorescence and for nuclei (blue) with DAPI. The experiment was performed in triplicate

Inhibition of CLEC4M-mediated transinfection with JFH-1 HCVcc by anti-CLEC4M antibody. Huh7-25-CLEC4M, -NC, and Huh7-it cells were pre-incubated with anti-CLEC4M antibody for 30 min before incubation with JFH-1 HCVcc for 4 h at 37 °C. After being washed, the cells were co-cultured with Huh7-it cells for 3 days, and HCV RNA was measured. As CD81-positive Huh7-it cells were permissive for HCV entry and highly susceptible to HCV replication, the level of HCV RNA 3 days post-incubation was 100-fold higher than in Huh7-25-CLEC4M cells. Bars indicate standard errors of triplicate experiments. Ctrl., immunoglobulin isotype control; *, P < 0.05
Discussion
In this study, we showed that CLEC4M was inactive in in vitro cisinfection but active in transinfection with HCVcc, and that the transinfection was mediated by a carbohydrate-recognition domain of CLEC4M. This was directly proven, for the first time, by using infectious HCVcc and particular host Huh7 cells (Huh7-25-CLEC4M) that were permissive for HCV RNA replication, negative for CD81, and positive for CLEC4M.
We directly showed that CLEC4M does not serve as an endocytotic receptor for HCV infectious particles or as a helper for HCV endocytosis in place of CD81. DC-SIGN, which is analogous to the CLEC4M protein, functions as an endocytotic receptor and has internalization motifs, such as a tri-acidic cluster, a dileucine motif and a tyrosine-based motif in the intracellular N-terminal domain [13]. The tri-acidic cluster and dileucine motif are present in the N-terminal region of CLEC4M, but the tyrosine-based motif is absent [13]. However, CLEC4M-positive Jurkat cells were susceptible to Ebola virus cisinfection [3]. CLEC4M-positive CHO cells captured SARS-CoV and incorporated, it leading to proteasome-dependent viral degradation [8]. CLEC4M-positive K562 and THP-1 cells internalized HCV virus-like particles consisting of recombinant envelope proteins E1 and E2, which differ from HCVpp [17]. Thus, the internalization by CLEC4M-positive cells is dependent on the type of cell and ligands. The early stage of infection such as attachment and entry of HCV requires particular cooperation of multiple receptors including coreceptors. Such an interaction between CLEC4M and other active receptors for HCV might be deficient in Huh7-25-CLEC4M cells.
To clarify physiological relevance of CLEC4M to HCV tropism, it is necessary to examine human liver sinusoidal cells (LSECs) for cis- and transinfection with HCVcc. Although we demonstrated that CLEC4M of human liver had a different glycosylation pattern from that of Huh7-25-CLEC4M cells (Fig. 1D), Ludwig and coworkers showed that HCV virus-like particles bound to LSECs of human liver tissue sections in situ [17]. Thus, a different glycosylation pattern of CLEC4M on LSECs may not affect the attachment of HCV. Lai and colleagues demonstrated that CLEC4M was expressed on human LSECs and functioned in binding HCV E2 glycoproteins but that primary human LSECs did not support HCVpp entry [15]. Thus, CLEC4M on LSECs is not an entry receptor for HCV. Therefore, LSECs should be resistant to HCVcc infection, and LSECs were indeed found to be resistant to HCVcc when co-cultured with J6/JFH-infected Huh-7.5 cells [15]. It is still unknown whether LSECs are permissive for HCV replication or not. Recently, Fletcher and associates showed that LSECs were not infected with HCVpp because expression of SR-BI and CLDN1 in the cells was very low [11]. They found that human brain endothelial cells expressed functional receptors that supported entry and replication of HCVcc. These results suggest that LSECs are unsusceptible to HCV infection due to the lower abundance of a few co-receptors on these cells. LSECs possibly function as a mediator of transinfection with HCV.
Several investigators have shown that CLEC4M mediates transinfection with HCVpp [9, 10, 16]. We showed in the present study that CLEC4M actually mediated transinfection with bona fide HCVcc. However, the attachment of HCVcc to Huh7-25-NC cells was apparently similar to that of Huh7-25-CLEC4M (Fig. 3A). Nonspecific binding may be responsible for the attachment to Huh7-25-NC cells, which is qualitatively different from that of Huh7-25-CLEC4M cells. Such nonspecific binding is probably too weak to mediate transinfection of HCVcc to adjacent Huh7-it cells. The lack of internalization in this case indicates that transinfection might occur through the release of captured HCVcc to adjacent hepatocytes. In the case of human liver in vivo, transinfection mediated by LSECs can occur with or without internalization. In duck liver in vivo, duck hepatitis B virus was captured by LSECs, internalized into LSECs and then transferred to adjacent hepatocytes [6]. HIV-1 was also incorporated into dendritic cells by DC-SIGN and then transmitted to CD4 T cells via exosomes [21]. Thus, these viruses were temporarily pooled in reservoir cells to escape from host immune evasion. It is still unknown whether CLEC4M on human LSECs internalizes HCV and whether human LSECs are permissive for HCV RNA replication.
In conclusion, we demonstrate that CLEC4M is a capture receptor for JFH-1 HCVcc but cannot substitute for CD81 as an entry receptor. Therefore, CLEC4M does not mediate cisinfection but does mediate transinfection without internalization of JFH-1 HCVcc. These findings and others suggest that CLEC4M on LSECs is responsible for the hepatotropism of HCV infection by capturing and releasing HCV to adjacent hepatocytes, but not by serving as a receptor for HCV cisinfection.
References
Agnello V, Abel G, Knight GB, Muchmore E (1998) Detection of widespread hepatocyte infection in chronic hepatitis C. Hepatology 28:573–584
Akazawa D, Date T, Morikawa K, Murayama A, Miyamoto M, Kaga M, Barth H, Baumert TF, Dubuisson J, Wakita T (2007) CD81 expression is important for the permissiveness of Huh7 cell clones for heterogeneous hepatitis C virus infection. J Virol 81:5036–5045
Alvarez CP, Lasala F, Carrillo J, Muniz O, Corbi AL, Delgado R (2002) C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J Virol 76:6841–6844
Appelmelk BJ, van Die I, van Vliet SJ, Vandenbroucke-Grauls CM, Geijtenbeek TB, van Kooyk Y (2003) Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J Immunol 170:1635–1639
Bashirova AA, Geijtenbeek TB, van Duijnhoven GC, van Vliet SJ, Eilering JB, Martin MP, Wu L, Martin TD, Viebig N, Knolle PA, KewalRamani VN, van Kooyk Y, Carrington M (2001) A dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN)-related protein is highly expressed on human liver sinusoidal endothelial cells and promotes HIV-1 infection. J Exp Med 193:671–678
Breiner KM, Schaller H, Knolle PA (2001) Endothelial cell-mediated uptake of a hepatitis B virus: a new concept of liver targeting of hepatotropic microorganisms. Hepatology 34:803–808
Burlone ME, Budkowska A (2009) Hepatitis C virus cell entry: role of lipoproteins and cellular receptors. J Gen Virol 90:1055–1070
Chan VS, Chan KY, Chen Y, Poon LL, Cheung AN, Zheng B, Chan KH, Mak W, Ngan HY, Xu X, Screaton G, Tam PK, Austyn JM, Chan LC, Yip SP, Peiris M, Khoo US, Lin CL (2006) Homozygous L-SIGN (CLEC4M) plays a protective role in SARS coronavirus infection. Nat Genet 38:38–46
Cormier EG, Durso RJ, Tsamis F, Boussemart L, Manix C, Olson WC, Gardner JP, Dragic T (2004) L-SIGN (CD209L) and DC-SIGN (CD209) mediate transinfection of liver cells by hepatitis C virus. Proc Natl Acad Sci USA 101:14067–14072
Falkowska E, Durso RJ, Gardner JP, Cormier EG, Arrigale RA, Ogawa RN, Donovan GP, Maddon PJ, Olson WC, Dragic T (2006) L-SIGN (CD209L) isoforms differently mediate trans-infection of hepatoma cells by hepatitis C virus pseudoparticles. J Gen Virol 87:2571–2576
Fletcher NF, Wilson GK, Murray J, Hu K, Lewis A, Reynolds GM, Stamataki Z, Meredith LW, Rowe IA, Luo G, Lopez-Ramirez MA, Baumert TF, Weksler B, Couraud PO, Kim KS, Romero IA, Jopling C, Morgello S, Balfe P, McKeating JA (2012) Hepatitis C virus infects the endothelial cells of the blood–brain barrier. Gastroenterology 142(634–643):e636
Ishibashi M, Wakita T, Esumi M (2010) 2′,5′-Oligoadenylate synthetase-like gene highly induced by hepatitis C virus infection in human liver is inhibitory to viral replication in vitro. Biochem Biophys Res Commun 392:397–402
Khoo US, Chan KY, Chan VS, Lin CL (2008) DC-SIGN and L-SIGN: the SIGNs for infection. J Mol Med 86:861–874
Koppel EA, Ludwig IS, Hernandez MS, Lowary TL, Gadikota RR, Tuzikov AB, Vandenbroucke-Grauls CM, van Kooyk Y, Appelmelk BJ, Geijtenbeek TB (2004) Identification of the mycobacterial carbohydrate structure that binds the C-type lectins DC-SIGN, L-SIGN and SIGNR1. Immunobiology 209:117–127
Lai WK, Sun PJ, Zhang J, Jennings A, Lalor PF, Hubscher S, McKeating JA, Adams DH (2006) Expression of DC-SIGN and DC-SIGNR on human sinusoidal endothelium: a role for capturing hepatitis C virus particles. Am J Pathol 169:200–208
Lozach PY, Amara A, Bartosch B, Virelizier JL, Arenzana-Seisdedos F, Cosset FL, Altmeyer R (2004) C-type lectins L-SIGN and DC-SIGN capture and transmit infectious hepatitis C virus pseudotype particles. J Biol Chem 279:32035–32045
Ludwig IS, Lekkerkerker AN, Depla E, Bosman F, Musters RJ, Depraetere S, van Kooyk Y, Geijtenbeek TB (2004) Hepatitis C virus targets DC-SIGN and L-SIGN to escape lysosomal degradation. J Virol 78:8322–8332
Pham TN, King D, Macparland SA, McGrath JS, Reddy SB, Bursey FR, Michalak TI (2008) Hepatitis C virus replicates in the same immune cell subsets in chronic hepatitis C and occult infection. Gastroenterology 134:812–822
Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM (2009) Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886
Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ (2005) Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796
Wu L, KewalRamani VN (2006) Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol 6:859–868
Zeisel MB, Fofana I, Fafi-Kremer S, Baumert TF (2011) Hepatitis C virus entry into hepatocytes: molecular mechanisms and targets for antiviral therapies. J Hepatol 54:566–576
Acknowledgments
The authors thank Dr. M. Oshima, National Institute of Infection Diseases, Tokyo, Japan, for providing us with Huh7-it cells. This work was supported by a grant for “Open Research Center” Project for Private Universities: matching fund subsidy from MEXT (2005), Nihon University Joint Research Grant 06-023, Grant-in-Aid for Scientific Research (C) 22590350 from MEXT (2010), and “Strategic Research Base Development” Program for Private Universities subsidized by MEXT (2010). English editorial assistance was provided by NAI Inc. and funded by Nihon University School of Medicine 50th Anniversary Fund Research Grant (2012).
Conflict of interest
All authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ishibashi, M., Morita, N., Nomura-Kawaguchi, C. et al. CLEC4M-positive and CD81-negative Huh7 cells are not susceptible to JFH-1 HCVcc infection but mediate transinfection. Arch Virol 159, 2949–2955 (2014). https://doi.org/10.1007/s00705-014-2150-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-014-2150-z