
Abstract
Pathological changes in corticobasal degeneration (CBD) consist of abnormal deposition of the microtubule-associated protein tau. However, the simultaneous accumulation of different misfolded proteins in the brain can be observed in many neurodegenerative diseases with significantly longer disease durations. We encountered a patient with CBD who survived for an extremely long period (18 years) after the diagnosis. We performed an autopsy to elucidate the effect of the longer survival on the pathology of CBD. We observed abnormal aggregation of trans-activating response region DNA-binding protein of 43 kDa (TDP-43) and α-synuclein, as well as phosphorylated tau, in neurons of broader regions of the brain, beyond the amygdala and other limbic areas. We found that phosphorylated tau, α-synuclein, and TDP-43 partially co-existed in the same cellular aggregates. The triple pathologic changes might be related to the longer survival of the patient compared with the typical clinical course of patients with CBD. Further investigations are required to support the hypothesis that tauopathy, synucleinopathy, and TDP-43 proteinopathy might share common pathogenic mechanisms in terms of cross-seeding of the pathologic proteins.
Similar content being viewed by others

Introduction
Corticobasal degeneration (CBD) is a sporadic neurodegenerative movement disorder first described by Rebeiz et al. [1]. CBD is clinically characterized by asymmetric akinesis and rigidity, upper limb dystonia, apraxia, myoclonus, and cognitive dysfunction [2]. Histopathological changes of CBD consist of tau pathology, which features abnormal deposition of the microtubule-associated protein tau, similar to progressive supranuclear palsy (PSP). CBD is pathologically characterized by neuropil threads in the gray and white matter, as well as astrocytic plaques and ballooned achromatic neurons [3–5].
Some patients afflicted by other neurodegenerative diseases with a different underlying pathology also present with similar clinical features as CBD [6]. Therefore, the term corticobasal syndrome (CBS) is commonly used to describe the clinical features, whereas the term CBD refers to the pathological entity [6]. The most frequent causes of CBS are CBD, followed by PSP, frontotemporal lobar degeneration (FTLD) with TAR-DNA-binding protein 43 (TDP-43) pathology, Pick’s disease, dementia with Lewy bodies (DLB), FTLD with fused in sarcoma/translated in liposarcoma (FUS/TLS) pathology, and Creutzfeldt–Jakob disease [6, 7]. Thus, compared to Parkinsonism syndromes, it is difficult to achieve accurate CBD diagnoses.
Analyses of the natural history and survival of CBD patients confirmed by postmortem examination has revealed that median survival time after symptom onset was 7.9 (range 2.5–12.5) years, and, after the first clinic visit, 4.9 (range 0.8–10) years [8]. However, we encountered a CBD patient whose disease was eventually confirmed biochemically and histologically. He survived for an extremely long period (18 years) after the initial diagnosis. An autopsy was performed to elucidate the potential effect of the longer survival on the CBD pathology. Surprisingly, we observed an unusual histological change in the broader central nervous system (CNS). Growing evidence suggests that the simultaneous accumulation of different misfolded proteins in the CNS could be observed in many neurodegenerative diseases with significantly longer disease durations [9]. To understand the relationship of the longer survival of a CBD patient and the concomitant aggregation of misfolded proteins in the CNS, we sought to determine whether phosphorylated tau, α-synuclein, and TDP-43 co-exist in the same cellular aggregates or in different brain regions and cell populations.
Patients and methods
Case report
The study was approved by the Ethics Committee of Kumamoto University Hospital. A 62-year-old Japanese male without family history of neurological diseases nor consanguineous history noticed clumsiness in the right upper and lower limbs in 1993, and subsequently reported difficulty in handwriting 3 years later (age 65 years). His clinical features slowly deteriorated and included gait disorder associated with postural instability and frequent falls, which caused head trauma in 1999. Upon his admission in 2000, neurological examination was performed; it revealed dysarthria, dysphagia, neck rigidity, asymmetric bradykinesia and rigidity, myoclonic or dystonic involuntary movement predominant in the right upper and lower limbs, impairment in two-point discrimination and skin writing sensation, moderate gait disturbance, and propulsion requiring assistance. Neuropsychological examination showed the presence of limb-kinetic apraxia, constructional and ideomotor apraxia, and poor word fluency, but no alien hand signs. Brain magnetic resonance imaging performed 5 years after onset (age 66 years) showed moderate cerebral atrophy, pronounced in the left precentral gyrus (supplementary Fig. 1a–c). The patient did not respond to levodopa or dopaminergic medications. He suffered from recurrent aspiration pneumonia in January 2001, and underwent permanent tracheostomy and subsequently tube feeding in March 2002. His condition gradually worsened, and he became comatose and presented with decorticate posture. In November 2010, respiratory failure with Cheyne–Stokes respiration and hypothermia developed. Brain computed tomography (CT) performed 18 years after CBD onset (age 79 years) demonstrated diffuse and severe brain atrophy with ventricular dilatation (supplementary Fig. 1d). His blood pressure gradually dropped, and he died in February 2011.

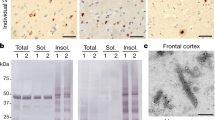
a The autopsied brain before fixation with formalin shows diffuse atrophy that was predominant in the left hemisphere, particularly the frontal and temporal lobes, with less atrophy in the parietal lobe. b The globus pallidus, subthalamic nucleus, and dentate nucleus are atrophic and brownish. c The substantia nigra is markedly depigmented. d Immunoblots of the sarkosyl-insoluble brain extracts before and after dephosphorylation derived from frontal (F), temporal (T), parietal lobe (P), and cerebellar (Cb) lesions. Blotting with the T46 antibody illustrates the typical doublet of 64 and 68 kDa (arrowheads) as well as two closely related bands of approximately 37 kDa (arrow), with the upper band slightly predominant over the lower one. In contrast, these bands were not observed in the brain extract from the cerebellar cortex. After dephosphorylation, the 64- and 68-kDa bands shifted to the same positions occupied by the 4R0 and 4R1 N bands, suggesting that four-repeat tau predominantly accumulated in our subject. e Immunoblots for phosphorylated TDP-43 (pS409/410) performed using brain extracts derived from frontal (F), temporal (T), parietal lobe (P), and cerebellar (Cb) lesions. High molecular weight smearing, phosphorylated full-length TDP-43 at 45 kDa (arrow), and several C-terminal fragments at 23, 24, and 26 kDa (arrowhead), with the 23-kDa band being the most intense, are detected and corresponded to FTLD-TDP type A. f Immunoblots for phosphorylated α-synuclein (pS129) performed using brain extracts derived from frontal (F), temporal (T), parietal lobe (P), and cerebellar (Cb) lesions showing faint bands in brain extracts derived from the frontal, temporal, and parietal lobes (arrowhead)
Sarkosyl-insoluble tau extraction and dephosphorylation
The procedures followed were in accordance with the standards of the Committee on Human Experimentation of Kumamoto University. Sarkosyl-insoluble tau and soluble tau were extracted from the frontal, temporal, and parietal lobes as well as the cerebellum of the patient. Dephosphorylation of insoluble tau was performed as described previously [10]. Tau proteins before and after dephosphorylation were electrophoresed on 10 % sodium dodecyl sulfate–polyacrylamide gels, transferred to polyvinylidene difluoride membranes, and probed with a phosphorylation-independent anti-tau mouse monoclonal antibody (T46, 1:1,000; Life Technologies, Carlsbad, CA, USA). Sarkosyl-insoluble, urea-soluble fractions extracted from each region were immunoblotted with the phosphorylation-dependent anti-TDP-43 rabbit polyclonal antibody (pS409/410, 1:1,000; Cosmo Bio, Tokyo, Japan) and phosphorylation-dependent anti-α-synuclein mouse monoclonal antibody (pS129, 1:2,000; Wako, Osaka, Japan).
Immunohistochemical analyses
For immunohistochemical studies, the following primary antibodies were used by standard avidin–biotin peroxidase methods: mouse anti-TDP-43 (Abnova, Taipei City, Taiwan), mouse anti-tau (Leica Biosystems, Wetzlar, Germany), and mouse anti-α-synuclein (Leica Biosystems, Buffalo Grove, IL, USA). For double labeling, fresh frozen tissue sections were fixed with 4 % paraformaldehyde and blocked with 5 % normal donkey serum/0.2 % Triton-X in phosphate-buffered saline. Combinations of rabbit anti-TDP-43 (1:500; Proteintech, Chicago, IL, USA) and mouse anti-phosphorylated tau (pS396, 1:250; Cell Signaling Technology, Beverly, MA, USA); or mouse anti-α-synuclein (1:250; BD Transduction Laboratories, Franklin Lakes, NJ, USA) were used. Immunolabeling was visualized using the Alexa 488-conjugated anti-mouse immunoglobulin antibody (1:200; Life Technologies) and Alexa 546-conjugated anti-rabbit immunoglobulin antibody (1:200; Life Technologies). Sections were examined using confocal microscopy (FV300, Olympus, Tokyo, Japan). The pathology was evaluated according to a semiquantitative scale: −, negative; +, occasional inclusions; ++, moderate inclusions; +++, frequent inclusions (Table 1).
Results
The autopsy was performed 11 h after death. The patient showed typical decorticate posture. The brain weighed 1,020 g and was diffusely atrophic. However, the atrophy was more predominant in the left hemisphere, particularly in the frontal and temporal lobe, with less observed in the parietal lobe (Fig. 1a). In cross-sections, the globus pallidus, subthalamic nucleus, and dentate nucleus were atrophic and brownish (Fig. 1b). The substantia nigra was markedly depigmented (Fig. 1c).
Using sarkosyl-insoluble brain extracts derived from the frontal, temporal, and parietal lobe, immunoblots (before dephosphorylation) stained with T46, an antibody that recognizes the C-terminal region of tau, illustrated the typical doublet of 64 and 68 kDa, as well as two closely related bands of approximately 37 kDa with a slight predominance of the upper band over the lower one (Fig. 1d). In contrast, these bands were not observed in the brain extracts from the cerebellar cortex (Fig. 1d). Immunoblotting performed after dephosphorylation showed that the 64- and 68-kDa bands had shifted to the same positions occupied by the 4R0 and 4R1 N bands, indicating that four-repeat tau predominantly accumulated in our subject. Immunoblot analyses using an antibody specific for abnormal TDP-43 (pS409/410) showed high molecular weight smearing, phosphorylated full-length TDP-43 at 45 kDa, and several C-terminal fragments at 23, 24, and 26 kDa, with the 23-kDa band the most intense (Fig. 1e). This result corresponds to the FTLD-TDP type A described by Tsuji et al. [11]. Immunoblots using anti-phosphorylated α-synuclein antibodies identified each of these faint bands in brain extracts derived from the frontal, temporal, and parietal lobe (Fig. 1f).
In the sections from formalin-fixed and paraffin-embedded tissues, ballooned neurons (Fig. 2a) and tau-positive astrocytic plaques with numerous neuropil threads predominantly in the frontotemporal lobes (Fig. 2b, c). Degenerative neurons were observed in the cortices, basal ganglia (Fig. 2d), an olivary nucleus, pontine nuclei, and cerebellar dentate nuclei with intracytoplasmic inclusions. The substantia nigra, as well as the basal ganglia showed moderate neuronal loss, and nearly all of the remaining pigmented neurons contained intracytoplasmic inclusions.

a Ballooned neuron in the parietal cortex. b, c Tau immunohistochemistry showing astrocytic plaques with numerous background tau-positive neuropil threads in the frontal (b) and parietal (c) cortex. d Degenerating neurons in the globus pallidus. Scale bars 20 μm (a, d) and 50 μm (b, c)
Immunohistochemical studies were performed using monoclonal antibodies against phosphorylated tau, α-synuclein, and TDP-43. The distribution of phosphorylated tau, TDP-43, and α-synuclein in neurons of the brain are summarized in Table 1. Immunohistochemical staining revealed that phosphorylated tau pathology was widely distributed in neurons of the brainstem nuclei (Fig. 3g, j, m), basal ganglia (Fig. 3p), as well as the cerebral cortices (Fig. 3a, d). Immunohistochemistry for TDP-43 showed mainly round-shaped neuronal cytoplasmic inclusions in the brainstem nuclei (Fig. 3h, k, n), cerebral cortices (Fig. 3b, e), and to a lesser extent, the basal ganglia (Fig. 3q). In the cerebral cortices, TDP-43 immunohistochemistry showed many short dystrophic neurites and neuronal cytoplasmic inclusions predominantly in the neocortical layer 2. Thus, the pattern of TDP-43 pathology was almost classified to FTLD-TDP type A designated by Mackenzie et al. [12]. α-synuclein-positive intraneuronal inclusions were distributed predominantly in the cerebral cortices (Fig. 3c, f), basal ganglia (Fig. 3r), and pontine nuclei (Fig. 3l). Interestingly, the distribution pattern of α-synuclein did not necessarily fit into the Braak stages for Lewy pathology [13].

Immunohistochemistry of phosphorylated tau (a, d, g, j, m, p), TDP-43 (b, e, h, k, n, q), and α-synuclein (c, f, i, l, o, r) in the temporal (a, b, c) and frontal cortices (d, e, f), substantia nigra (g, h, i), pontine nuclei (j, k, l), medulla oblongata (m, n, o), and globus palidus (p, q, r). Scale bars 20 μm. P-tau phosphorylated tau, α-syn α-synuclein
We assessed whether a possible synergism existed among TDP-43, phosphorylated tau, and α-synuclein. Immunostaining for phosphorylated tau demonstrated round neuronal cytoplasmic inclusions in the frontal cortex (Fig. 4a, d). Interestingly, the phosphorylated tau-positive inclusions frequently co-localized with TDP-43 (Fig. 4a–f). In contrast, α-synuclein-positive neuronal intracytoplasmic inclusions occasionally intermingled with TDP-43 (Fig. 4g–i). Approximately 57 and 34 % of TDP-43-positive neuronal cytoplasmic inclusions co-existed with phosphorylated tau or α-synuclein, respectively.

a–f Confocal microscopy analyses of the localization of phosphorylated tau (a, d) and pan-TDP-43 (b, e) in the frontal cortex. Merged images are presented in (c, f). g–i Confocal microscopy analyses of the localization of α-synuclein (g) and pan-TDP-43 (h) in the frontal cortex. The merged image is presented in (i). Arrows indicate neuronal cytoplasmic inclusions stained with anti-phosphorylated tau or α-synuclein antibodies. Arrowheads show neuronal cytoplasmic inclusions stained with anti-TDP-43 antibody. Nuclei were stained with 4′,6-diamidino-2-phenylindole. Scale bars 50 μm
Discussion
In this report, we hypothesize that the extremely long survival of a patient with CBD might result in the triple pathology of α-synuclein, phosphorylated tau, and TDP-43 proteinopathy.
Both PSP and CBD are characterized by intracytoplasmic aggregates of hyperphosphorylated tau with four microtubule-binding repeats. However, different proteolytic processing of abnormal tau has been found in these two diseases based on immunoblots of sarkosyl-insoluble brain extracts: a 33-kDa band predominates in the low molecular weight tau fragments in PSP, whereas two closely related bands of approximately 37 kDa predominate in CBD [10]. Our data from immunoblots of sarkosyl-insoluble brain extracts showed the specific proteolytic pattern of abnormal tau consistent with CBD, although phosphorylated tau pathology was widely distributed in the brainstem nuclei, basal ganglia, and cerebral cortices as well as astrocytic plaques. Thus, our CBD diagnosis of the patient has been proven biochemically and histologically.
The concomitant accumulation of various proteins is a common feature in many neuromuscular degenerative diseases. The co-accumulation of tau and α-synuclein in patients with α-synuclein-related diseases has been shown in familial and sporadic PD, DLB, and multiple system atrophy [14–22]. Lewy bodies with widespread tau and alpha-synuclein deposition were specifically observed in the limbic areas of DLB brains, mainly associated with amyloid-β (Aβ) deposits [15, 17–20, 23]. In contrast, the concomitant deposition of α-synuclein and tau has been detected in patients with tau-related diseases, such as familial and sporadic Alzheimer’s disease (AD), Down syndrome, PSP, Parkinsonism-dementia complex of Guam, and frontotemporal dementia (FTD) [24–31]. Elsewhere, co-localization of tau and α-synuclein was typically limited to the amygdala and other limbic areas in these cases [32]. To our knowledge, our CBD patient is the first case in which concomitant deposition of α-synuclein and tau in broader CNS regions was confirmed biochemically and histologically. The study of TDP-43 pathology in tauopathy patients, including CBD, demonstrated predominantly glial TDP-43 pathology with staining of tau-immunoreactive astrocytic plaque-like structures and coiled bodies in 15.4 % of CBD cases [9]. To the best of our knowledge, this is the first report to show the abnormal aggregation of phosphorylated tau, TDP-43, and α-synuclein in neurons anywhere, beyond the amygdala and other limbic areas.
There are several in vitro studies suggesting pathological synergistic effects between α-synuclein and tau. α-synuclein aggregation could be greatly enhanced upon co-incubation with tau in a concentration-dependent manner in vitro [18]. A recent study demonstrated that tau co-localized and interacted with α-synuclein aggregates in H4 cells and primary neuronal cultures, and that the interaction was associated with more α-synuclein aggregates, high molecular weight species, and enhanced toxicity [22]. Moreover, a few fibrillized α-synuclein seeds have been shown to induce intracellular massive hyperphosphorylated tau aggregation, mimicking neurofibrillary tangles [22]. Thus, our findings might suggest that tau, TDP-43, and α-synuclein promote the aggregation of each other.
Mooney et al. [33] reported a rare case of a patient with co-occurrence of the pathologies of CBD and PD. Our patient chiefly demonstrated cortical apraxia with minor symptoms of Parkinsonism and dementia. Pathological evaluation revealed degeneration in the substantia nigra and basal ganglia, particularly the putamen and globus pallidus, followed by the caudate nuclei. Therefore, the possibility of chance association between tauopathy and synucleinopathy is noted, but it is inconsistent with lack of responsiveness to levodopa treatment. Kouri et al. [34] identified three cases of CBD with olivopontocerebellar atrophy, which was referred to as CBD-OPCA. The neuropathologic features showed no α-synuclein-positive glial cytoplasmic inclusions, but marked neuronal loss in pontine nuclei, inferior olivary nucleus, and Purkinje cell layer, as well as TDP-43 immunoreactive neuronal and glial cytoplasmic inclusions and threads throughout the basal ganglia and in olivopontocerebellar system. More precise histological analyses would be useful to know whether our case belongs to the same disease entity as CBD-OPCA.
Recent reviews illustrated that synergistic effects of tau protein, Aβ, α-synuclein and other pathologic proteins may represent a common final pathway leading to or preventing neuronal damage, and hypothesized that prion-like induction and spreading of these pathologic proteins are major pathophysiological mechanisms in various neurodegenerative diseases [35, 36]. In our case, the triple pathology of tauopathy, synucleinopathy, and TDP-43 proteinopathy may have contributed to the longer survival. Further investigations are required to support the hypothesis that tauopathy, synucleinopathy, and TDP-43 proteinopathy share common pathogenic mechanisms in terms of cross-seeding of pathologic proteins.
References
Rebeiz JJ, Kolodny EH, Richardson EP Jr (1968) Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol 18:20–33
Boeve BF, Lang AE, Litvan I (2003) Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 54(Suppl 5):S15–S19
Dickson DW, Bergeron C, Chin SS et al (2002) Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61:935–946
Feany MB, Dickson DW (1995) Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 146:1388–1396
Kouri N, Murray ME, Hassan A et al (2011) Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or Richardson syndrome. Brain 134:3264–3275
Ling H, O’Sullivan SS, Holton JL et al (2010) Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 133:2045–2057
Aiba I (2012) Corticobasal syndrome: recent advances and future directions. Brain Nerve 64:462–473
Wenning GK, Litvan I, Jankovic J et al (1998) Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry 64:184–189
Uryu K, Nakashima-Yasuda H, Forman MS et al (2008) Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol 67:555–564
Arai T, Ikeda K, Akiyama H et al (2004) Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol 55:72–79
Tsuji H, Arai T, Kametani F et al (2012) Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 135:3380–3391
Mackenzie IR, Neumann M, Baborie A et al (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122:111–113
Braak H, Del Tredici K, Rub U et al (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Galpern WR, Lang AE (2006) Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol 59:449–458
Clarimon J, Molina-Porcel L, Gomez-Isla T et al (2009) Early-onset familial Lewy body dementia with extensive tauopathy: a clinical, genetic, and neuropathological study. J Neuropathol Exp Neurol 68:73–82
Duda JE, Giasson BI, Mabon ME et al (2002) Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol 104:7–11
Galloway PG, Grundke-Iqbal I, Iqbal K, Perry G (1988) Lewy bodies contain epitopes both shared and distinct from Alzheimer neurofibrillary tangles. J Neuropathol Exp Neurol 47:654–663
Giasson BI, Forman MS, Higuchi M et al (2003) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300:636–640
Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW (2003) Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 62:389–397
Terni B, Rey MJ, Boluda S et al (2007) Mutant ubiquitin and p62 immunoreactivity in cases of combined multiple system atrophy and Alzheimer’s disease. Acta Neuropathol 113:403–416
Wills J, Jones J, Haggerty T et al (2010) Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp Neurol 225:210–218
Badiola N, de Oliveira RM, Herrera F et al (2011) Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS One 6:e26609
Iseki E, Marui W, Kosaka K, Ueda K (1999) Frequent coexistence of Lewy bodies and neurofibrillary tangles in the same neurons of patients with diffuse Lewy body disease. Neurosci Lett 265:9–12
Hamilton RL (2000) Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol 10:378–384
Lippa CF, Fujiwara H, Mann DM et al (1998) Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol 153:1365–1370
Raghavan R, Khin-Nu C, Brown A et al (1993) Detection of Lewy bodies in Trisomy 21 (Down’s syndrome). Can J Neurol Sci 20:48–51
Lippa CF, Schmidt ML, Lee VM, Trojanowski JQ (1999) Antibodies to alpha-synuclein detect Lewy bodies in many down’s syndrome brains with Alzheimer’s disease. Ann Neurol 45:353–357
Judkins AR, Forman MS, Uryu K et al (2002) Co-occurrence of Parkinson’s disease with progressive supranuclear palsy. Acta Neuropathol 103:526–530
Forman MS, Schmidt ML, Kasturi S et al (2002) Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol 160:1725–1731
Wilhelmsen KC, Forman MS, Rosen HJ et al (2004) 17q-linked frontotemporal dementia-amyotrophic lateral sclerosis without tau mutations with tau and alpha-synuclein inclusions. Arch Neurol 61:398–406
Yancopoulou D, Xuereb JH, Crowther RA, Hodges JR, Spillantini MG (2005) Tau and alpha-synuclein inclusions in a case of familial frontotemporal dementia and progressive aphasia. J Neuropathol Exp Neurol 64:245–253
Schmidt ML, Martin JA, Lee VM, Trojanowski JQ (1996) Convergence of Lewy bodies and neurofibrillary tangles in amygdala neurons of Alzheimer’s disease and Lewy body disorders. Acta Neuropathol 91:475–481
Mooney T, Tampiyappa A, Robertson T et al (2011) Dual pathology of corticobasal degeneration and Parkinson’s disease in a patient with clinical features of progressive supranuclear palsy. Neurol India 59:887–890
Kouri N, Oshima K, Takahashi M et al (2013) Corticobasal degeneration with olivopontocerebellar atrophy and TDP-43 pathology: an unusual clinicopathologic variant of CBD. Acta Neuropathol 125:741–752
Jellinger KA (2011) Interaction between alpha-synuclein and other proteins in neurodegenerative disorders. Sci World J 11:1893–1907
Jellinger KA (2012) Interaction between pathogenic proteins in neurodegenerative disorders. J Cell Mol Med 16:1166–1183
Acknowledgments
We would like to thank Ms. Mika Oka and the members of Department of Neurology, Kumamoto University Hospital for technical help with autopsy analyses and clinical data collection. This work was supported by a Grant-in-Aid for Scientific Research, the Ministry of Education, Culture, Sports, Science and Technology of Japan, and Grants-in-Aid from the Amyloidosis Research Committee, the Ministry of Health, Labour and Welfare of Japan.
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical standard
This study has been approved by the appropriate ethics committee and has therefore been performed in the accordance with the ethical standards laid down in the 1964 Declaration of Helsinki. The spouse of the patient gave informed consent instead of the patient.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Yamashita, N. Sakashita and T. Yamashita authors contributed equally to the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yamashita, S., Sakashita, N., Yamashita, T. et al. Concomitant accumulation of α-synuclein and TDP-43 in a patient with corticobasal degeneration. J Neurol 261, 2209–2217 (2014). https://doi.org/10.1007/s00415-014-7491-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-014-7491-8